Integrated Network Ethnopharmacology, Molecular Docking, and ADMET Analysis Strategy for Exploring the Anti-Breast Cancer Activity of Ayurvedic Botanicals Targeting the Progesterone Receptor
Asma Mokashi1 and Neela M. Bhatia1,*![]()
1Bharati Vidyapeeth College of Pharmacy, Kolhapur, Maharashtra, India
*Correspondence to: Neela M. Bhatia, Department of Quality Assurance, Bharati Vidyapeeth College of Pharmacy, Near Chitranagari, Kolhapur – 416013, Maharashtra, India, Tel: 9823463687. E-mail: neela.bhatia20@gmail.com
Received: August 11 2024; Revised: September 30 2024; Accepted: October 7 2024; Published Online: October 23 2024
Cite this paper:
Mokashi A, Bhatia NM. Integrated Network Ethnopharmacology, Molecular Docking, and ADMET Analysis Strategy for Exploring the Anti-Breast Cancer Activity of Ayurvedic Botanicals Targeting the Progesterone Receptor. BIO Integration 2024; 5: 1–17.
DOI: 10.15212/bioi-2024-0066. Available at: https://bio-integration.org/
Download citation
© 2024 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Background: In women, breast cancer is currently among the most common cancers and the second major cause of cancer-related mortality. One therapeutic target for breast cancer is the progesterone receptor (PR), which can be inhibited by specific PR modulators.
Methods: Current anti-cancer medications have notorious adverse effects. Consequently, an urgent need exists to identify less hazardous, more effective medicines with few to no adverse effects. One strategy uses ancient herbal remedies to create medications derived from nature. Herein, we used data from the Dr. Duke, IMPPAT, PubChem, Binding DB, UniProt, and DisGeNET databases to construct a network in Cytoscape 3.10.0. Through a polypharmacology approach, bioactives with similarity indices greater than 0.6 were screened and docked with the PR. The top ten ligands with good docking scores were further subjected to interaction analysis in AutoDock v.4.2 software. We additionally analyzed the ADMET properties of the phytochemicals.
Results: Procurcumenol and alpha-turmerone exhibited superior interactions with PR, with binding affinities of −7.85 kcal/mol. All compounds met Lipinski’s rule of five and were effective ligands for the PR according to ADMET data analysis. Our findings suggest that procurcumenol and alpha-turmerone may serve as potential anti-breast cancer agents; specifically targeting the PR in breast cancer cells.
Conclusion: Understanding of anti-breast cancer activity can be facilitated through experimental validation of network analysis and molecular docking findings.
Keywords
Ayurvedic botanicals, breast cancer, molecular docking, network pharmacology, progesterone receptor.
Introduction
Cancer remains a leading cause of mortality worldwide, ranking second only to cardiovascular disorders [1]. By 2040, 28.4 million new cancer cases have been projected annually, representing a 47% increase since 2020 [2]. Currently, breast cancer accounts for 2.3 million new cases worldwide, surpassing lung cancer as the most frequently diagnosed cancer. It continues to substantially influence global cancer-related mortality [3]. Each year, breast cancer affects approximately 1 million women and leads to more than 410,000 deaths [4]. By 2030, breast cancer is expected to be the deadliest cancer worldwide, with an estimated 10 million new cases [5].
Traditional treatments for breast cancer, including radiotherapy, surgery, and chemotherapy, have several drawbacks, such as toxicity and adverse effects [6–8]. Although numerous chemotherapeutic drugs are available, their effectiveness is limited by cancer cell diversity, the emergence of drug-resistant cell populations, and complex tumor interactions [9]. Consequently, researchers have explored novel phytonutrients and herbal materials as potential treatments [10].
Ayurvedic medicine, one of the oldest traditional healing systems, has long been recognized for its holistic approach to health and disease management. Research is increasingly focusing on the potential of Ayurvedic botanicals in cancer treatment, particularly because of their diverse bioactive compounds that exhibit anti-tumor properties. These naturally occurring compounds offer promising alternative treatments due to their wide availability, therapeutic potential, and reduced cytotoxicity [11]. These compounds have been found to inhibit various cancer development mechanisms, including tumor-promoting angiogenesis, replicative immortality, invasion, and metastasis [12]. Effective cancer treatment involves the targeted delivery of medications to diseased tissues while minimizing harm to healthy tissues [13]. Cancer cells often upregulate growth- and survival-promoting receptors on their surfaces; therefore, these receptors have provided reliable therapeutic targets [14, 15].
Breast cancer notably shows diverse receptor expression patterns: the ovarian steroid hormones progesterone and estrogen play key roles in disease progression through their respective receptors, the progesterone receptor (PR) and estrogen receptor (ER) [16–19]. Approximately 70% of breast cancers in older women are ER+/PR+, thus highlighting the potential for therapies targeting these receptors [17, 20, 21]. PGR, a member of the nuclear receptor superfamily, regulates the expression of genes associated with cell proliferation and differentiation in breast tissue. Its activation has been shown to influence tumor growth and metastasis; therefore, it is a crucial target for therapeutic intervention. These findings highlight the importance of investigating novel compounds that modulate this receptor, to contribute to more effective strategies for breast cancer management.
Numerous Ayurvedic botanicals targeting the PR have been tested for breast cancer treatment. Specifically, the structure-based multitargeted molecular docking analysis of selected furanocoumarins has shown promising interactions with breast cancer targets [21]. Additionally, molecular docking, as well as absorption, distribution, metabolism, excretion, and toxicity (ADMET) studies, have predicted the anti-breast cancer effects of aloin through targeting both the ER and PR [22]. In silico studies have further explored various phytochemicals—particularly 4-methoxy coumesterol, apigenin, biochanin, coumesterol, crocetin, curcumin, daidzein, diosgenin, formononetin, gabridin, genestein, hesperetin, indole-3-carbinol, kaempferol, lignan, luteolin, lycopene, naringenin, quercetin, and resveratrol—as anticancer agents, by evaluating their interactions with the ER and PR [23]. Moreover, recent research has highlighted the importance of virtual screening and molecular dynamics simulations to identify and optimize anti-breast cancer compounds from natural sources [24, 25].
Despite the wealth of available information, a gap remains in understanding the specific molecular mechanisms through which these botanicals interact with key targets, such as the PR, in breast cancer. Consequently, structure-based virtual screening is increasingly performed in medicinal research, because of its high efficiency [26]. Molecular docking is an affordable and effective method for predicting drug-receptor interactions and stable binding sites [27, 28]. Network pharmacology systematically accumulates and analyzes vast amounts of knowledge regarding phytochemicals, cellular targets, and mechanisms, thereby providing valuable insights into traditional medicines and improving current drugs’ reliability and effectiveness [29].
This study presents an innovative approach to evaluating the anti-breast cancer potential of selected Ayurvedic botanicals by integrating network ethnopharmacology and advanced molecular docking techniques. Our aim was to use a comprehensive in silico analysis to elucidate the complex molecular interactions between these botanicals and the PR, thus providing a deeper understanding of their therapeutic mechanisms. This integrative method not only highlights the synergy between traditional knowledge and modern computational tools, but also offers novel insights into the potential of these botanicals as effective agents against breast cancer.
Materials and methods
Network pharmacology studies
Screening of active ingredients
In this investigation, we examined the phytoconstituents from selected Ayurvedic botanicals (Withania somnifera, Asparagus racemosus, Azadirachta indica, Linum usitatissimum, Trigonella foenum-graecum, Curcuma longa, Aloe barbadensis, Glycyrrhiza glabra, and Murraya koenigii) traditionally documented to possess anti-breast cancer activity. Dr. Duke’s Phytochemical and Ethnobotanical Databases online platform [30], Indian Medicinal Plants, Phytochemistry and Therapeutics 2.0 [31], and literature mining were used to gather information regarding the phytoconstituents of selected Ayurvedic botanicals.
The investigation used data in .sdf format, which are available free of charge, and 3D structures of phytoconstituents. In PubChem [32], we looked up the common names and precise structures of the phytoconstituents of selected Ayurvedic botanicals.
Establishment of the target
The RCSB PDB database [33] was used to gather data on the PR. The species were confined to human sources, and the target was identified. The PR as a therapeutic target for breast cancer was searched with DisGeNET [34]. Through UniProt [35], a standard name for the protein target was found.
Prediction of bioactives targeting the PR
The .sdf files containing the structures of phytoconstituents from selected Ayurvedic botanicals were uploaded to the Binding DB (https://www.bindingdb.org) to predict the binding of bioactives targeting the PR for the treatment of breast cancer. Bioactives with a score between 0.6 and 1 were selected. The multiple databases linked to Binding DB were used to extract additional data on the target. The UniProt IDs provided in Binding DB were used to retrieve the protein symbols from UniProt. DisGeNET was searched for associations between the bioactive target and breast cancer.
Network construction
Cytoscape 3.10.0 was used to visually represent the network, and analyze and update the data. Data pairs of selected Ayurvedic botanicals with bioactive PCIDs, bioactive PCIDs with the PR, and the PR with breast cancer were built in Microsoft Excel. The data pairs were imported and used to create a network map of the therapeutic components and the disease target. The nodes in the network diagram indicate selected Ayurvedic botanicals, bioactives, PR, and breast cancer, whereas the edges indicate how the nodes are connected. The network was examined with the Network Analyzer function [36, 37].
Docking studies
Selection of ligands
Through a network pharmacology approach, we screened bioactives with a similarity index greater than 0.6 and that were found to be associated with the PR (1SQN) for further study.
Preparation of ligands
The 3D structures of inhibitors and their respective PubChem CIDs were obtained and saved in .sdf format. Furthermore, the 3D structures of all ligands were loaded into PyMOL software for conversion of 3D structures from .sdf to .pdb format. In PyMOL software, metals were also removed from the ligand structures for appropriate docking studies. The prepared ligands were saved in .pdb format. A number of torsions from zero to six was chosen; when any ligand showed more than six torsions, the value was adjusted to six. Hydrogen (H-) bond interactions were also calculated and described. The presence of H-bonds indicates stable interactions between ligands and proteins. Discovery Studio 2020 Client and Chimera software was used to depict H-bonds, 2D images, and protein-ligand interactions images for good visualization of the docking.
Preparation of the protein
The crystal structure of the target protein was retrieved from the Protein Data Bank (PDB) under PDB ID 1SQN and subjected to further docking studies.
Induced-fit molecular docking
Molecular docking, an important component of computer-assisted drug discovery, aids in predicting the intermolecular frameworks formed between proteins and ligands, and outputs the appropriate binding between molecules. Docking was performed in the 4.2.6 program, with the implemented empirical free energy function and the Lamarckian Genetic Algorithm. The grid maps were calculated with AutoGrid. In all dockings, a grid map with 40 × 40 × 40 points and a grid-point spacing of 0.714 Å was applied.
The best conformation with the lowest docked energy was chosen from the docking search. The interactions of complex protein-ligand conformations including H-bonds and bond lengths were analyzed in PyMOL software, UCSF Chimera, and Accelrys Discovery Studio Visualizer software [36, 38, 39].
CASTp identification
Computed Atlas of Surface Topography of Proteins (CASTp) (http://cast.engr.uic.edu) provides an online resource for locating, delineating, and measuring concave surface regions on 3D structures of proteins. These regions include pockets located on protein surfaces and voids buried in the protein interior [40].
ADMET studies
Computational ADMET prediction is frequently used in modern drug discovery to predict drug pharmacokinetics and toxicity. ADMET properties are necessary for the selection and development of drug candidates. The ADMET properties for the compounds alpha-atlantone, 18-alpha-glycyrrhetinic acid, alpha-turmerone, 18-beta-glycyrrhetinic acid, bisabola-3,10-dien-2-one, caffeic acid, curdione, curcumenone, dehydrocurdione, and procurcumenol were estimated in ADMETlab. Greater human intestinal absorption (HIA) indicates that a compound is better absorbed from the intestinal tract after oral administration. However, determining the toxicity of chemical compounds is necessary to identify their harmful effects on humans, animals, plants, or the environment [41]. Here, we used ADMETlab2.0 https://admetmesh.scbdd.com/ to verify the prediction of ADMET properties.
Results and discussion
Network pharmacology studies
Screening of active ingredients
The botanical ashwagandha (Withania somnifera) has been documented to contain 52 phytoconstituents. Shatavari (Asparagus racemosus) contains 28 phytoconstituents. Neem (Azadirachta indica) contains 23 phytoconstituents. Alsi seeds (Linum usitatissimum) contain 60 phytoconstituents. Methi (Trigonella foenum-graecum) contains 65 phytoconstituents. Haldi (Curcuma longa) contains 98 phytoconstituents. Ghritkumari (Aloe barbadensis) contains 21 phytoconstituents. Yashtimadhu (Glycyrrhiza glabra) contains 100 phytoconstituents. Finally, Kadi Patta (Murraya koenigii) contains 86 phytoconstituents.
Establishment of the target
For the PR, the data were as follows: PDB ID, progesterone receptor (1SQN); protein name, progesterone receptor; organism, Homo sapiens; resolution, 1.45 Å; sequence length = 261; UniProt ID P06401; gene name, HUMPR.
Prediction of bioactives against the target
A total of 32 bioactives from Ashwagandha (Withania somnifera), 3 from Shatavari (Asparagus racemosus), 2 from Neem (Azadirachta indica), 2 from Alsi seeds (Linum usitatissimum), 12 from Methi (Trigonella foenum-graecum), 10 from Haldi (Curcuma longa), 5 from Ghritkumari (Aloe barbadensis), 14 from Yashtimadhu (Glycyrrhiza glabra), and 4 from Kadi Patta (Murraya koenigii) showed interaction scores with the PR equal to or greater than 0.6 (Figure 1). The bioactives from selected Ayurvedic botanicals screened through a polypharmacology approach were subjected to network construction and analysis in Cytoscape v.3.2.1 software.
Figure 1 PPI network illustrating the interactions between bioactive compounds from selected Ayurvedic botanicals and the PR in the context of breast cancer. The network map visually represents the connections between key bioactives and PR, thereby highlighting potential therapeutic targets and pathways influenced by these compounds, which may contribute to their anti-breast cancer activity.
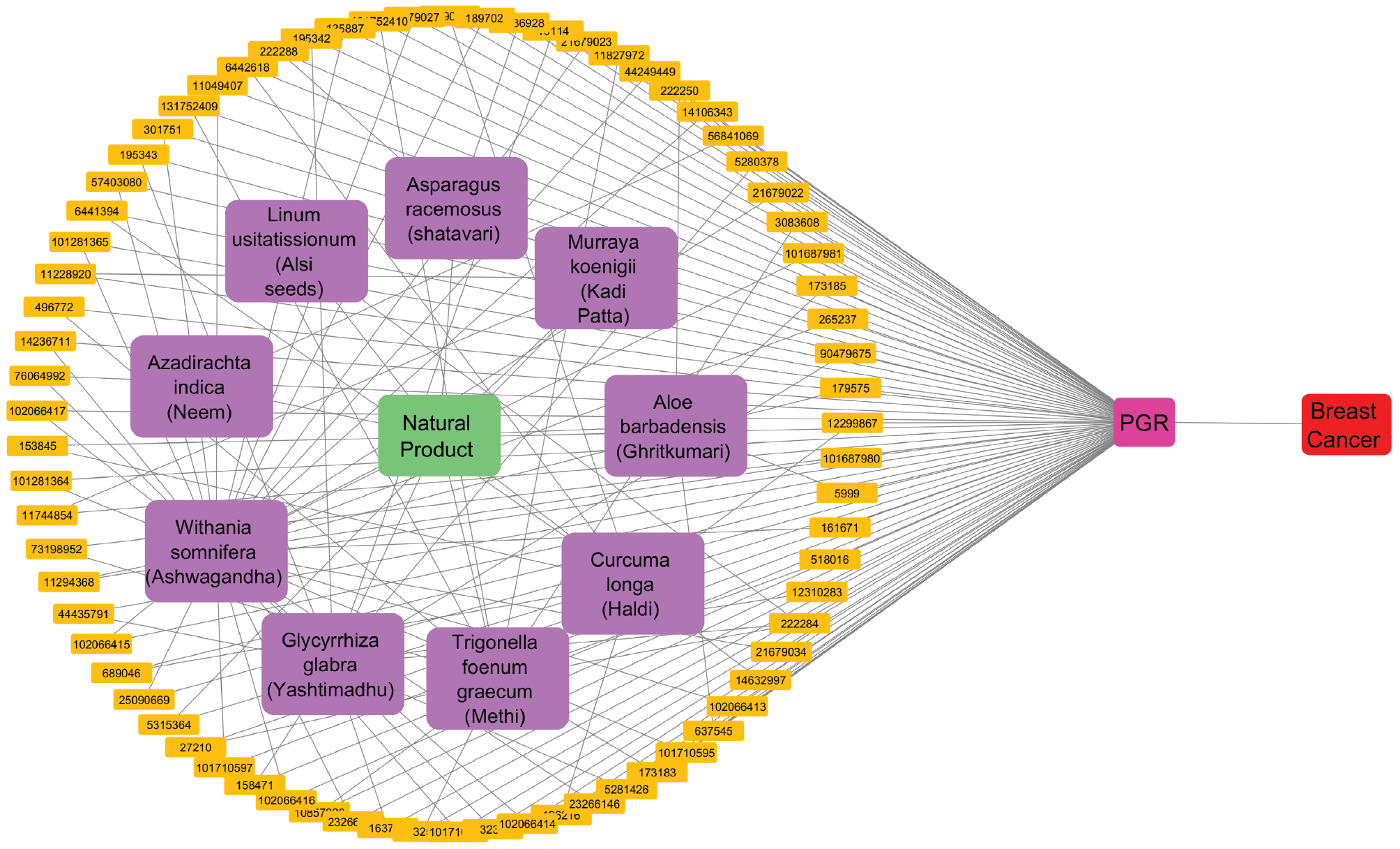
Network construction
The screened bioactives from selected Ayurvedic botanicals were subjected to network construction (Figure 1) and analysis in Cytoscape software.
Molecular docking studies
The process of identifying active site residues and targeting structures is crucial in drug design, particularly through ligand-protein docking. By predicting the active site and selecting the optimal 3D structure of the target protein, potential binding interactions can be better understood. In this study, docking was performed by using the PR with various ligands, and the conformations with the lowest docked energy were selected. The active site was predicted after selection of the 3D structure of the target protein. The best conformation with the lowest docked energy was chosen from the docking search. After docking of the PR (1SQN) with alpha-atlantone, 18-alpha-glycyrrhetinic acid, alpha-turmerone, 18-beta-glycyrrhetinic acid, bisabola-3,10-dien-2-one, caffeic acid, curdione, curcumenone, dehydrocurdione, and procurcumenol, we observed favorable binding energy between the protein and ligands for both alpha-turmerone and procurcumenol (−7.85 kcal/mol). A number of torsions from zero to six was chosen; if any ligand showed more than six torsions, the value was adjusted to six. H-bond interactions were also calculated and described. The presence of H-bonds indicates stable interactions between ligands and proteins. Discovery Studio 2020 Client and Chimera software were used to depict H-bonds, 2D images, and protein-ligand interaction images for good visualization of the docking. After docking of the PR (1SQN) with alpha-atlantone (−7.69 kcal/mol) (Figure 2), 18-alpha-glycyrrhetinic acid (−6.36 kcal/mol) (Figure 3), alpha-turmerone (−7.85 kcal/mol) (Figure 4), 18-beta-glycyrrhetinic acid (−5.38 kcal/mol) (Figure 5), bisabola-3, 10-dien-2-one (−7.56 kcal/mol) (Figure 6), caffeic acid (−5.87 kcal/mol) (Figure 7), curdione (−7.71 kcal/mol) (Figure 8), curcumenone (7.77 kcal/mol) (Figure 9), dehydrocurdione (7.73 kcal/mol) (Figure 10), and procurcumenol (7.85 kcal/mol) (Figure 11), we observed favorable binding energies between the protein and ligands (Table 1). The compounds 18-alpha-glycyrrhetinic acid and caffeic acid formed three hydrogen bonds with the PR.
Figure 2 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with alpha-atlantone shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
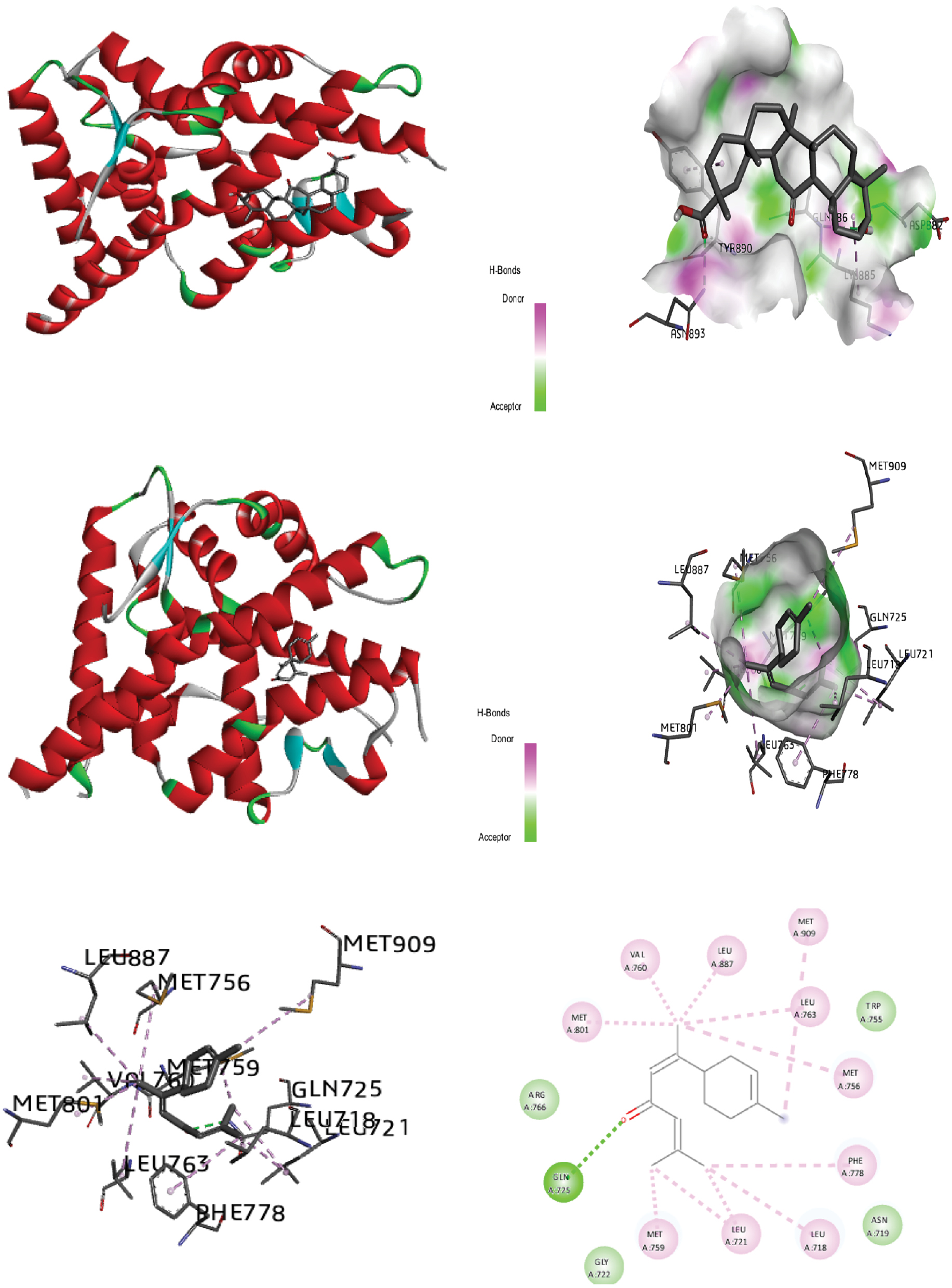
Figure 3 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with 18-alpha-glycyrrhetinic acid shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
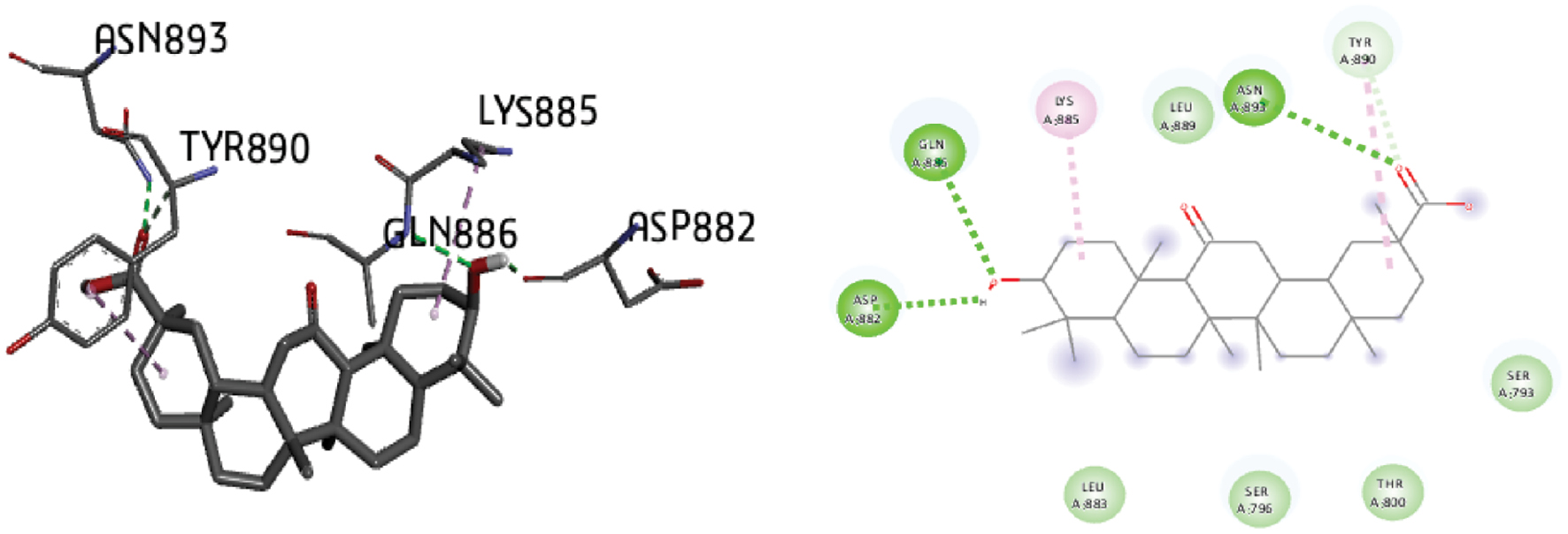
Figure 4 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with alpha-turmerone shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
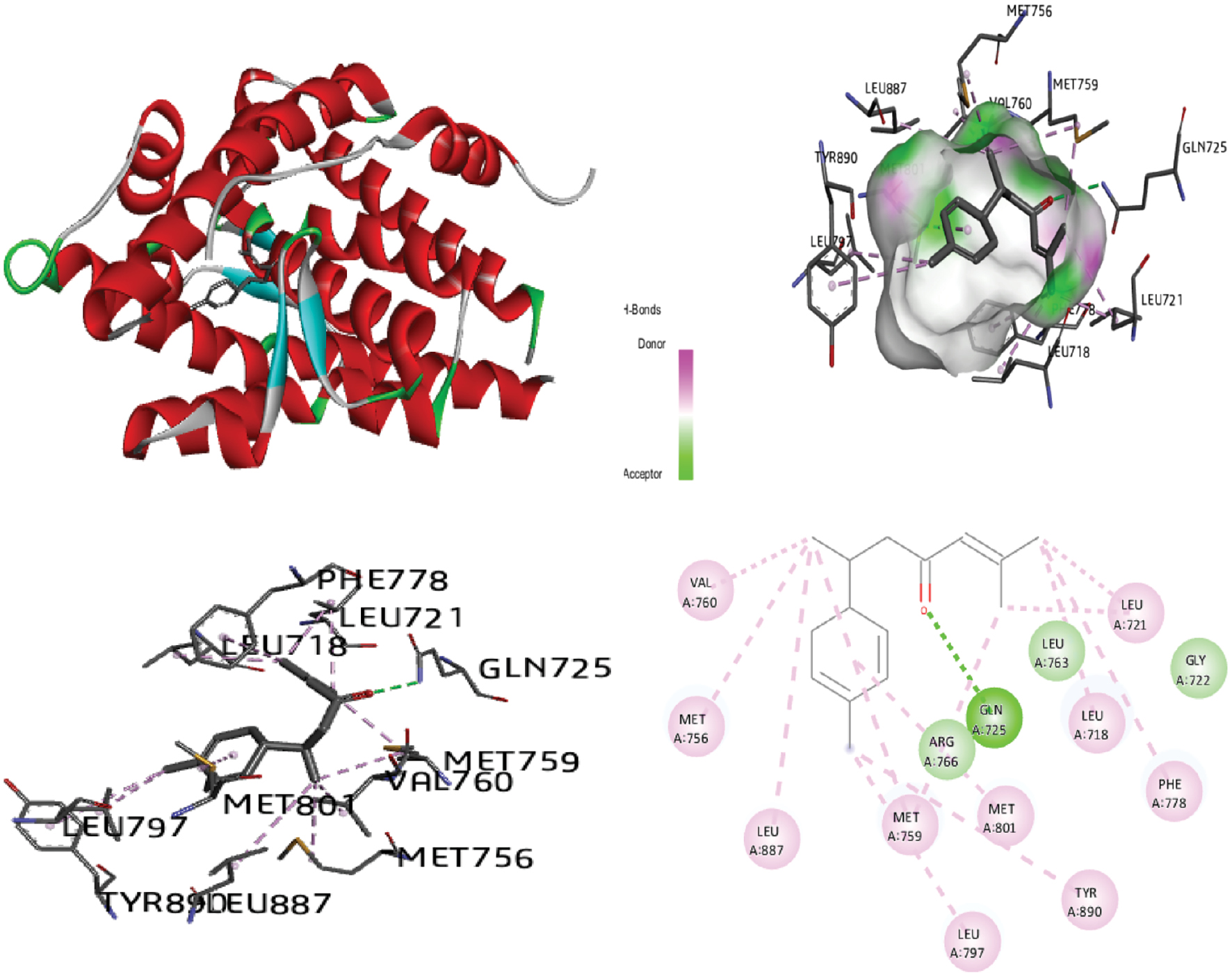
Figure 5 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with 18-beta-glycyrrhetinic acid shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
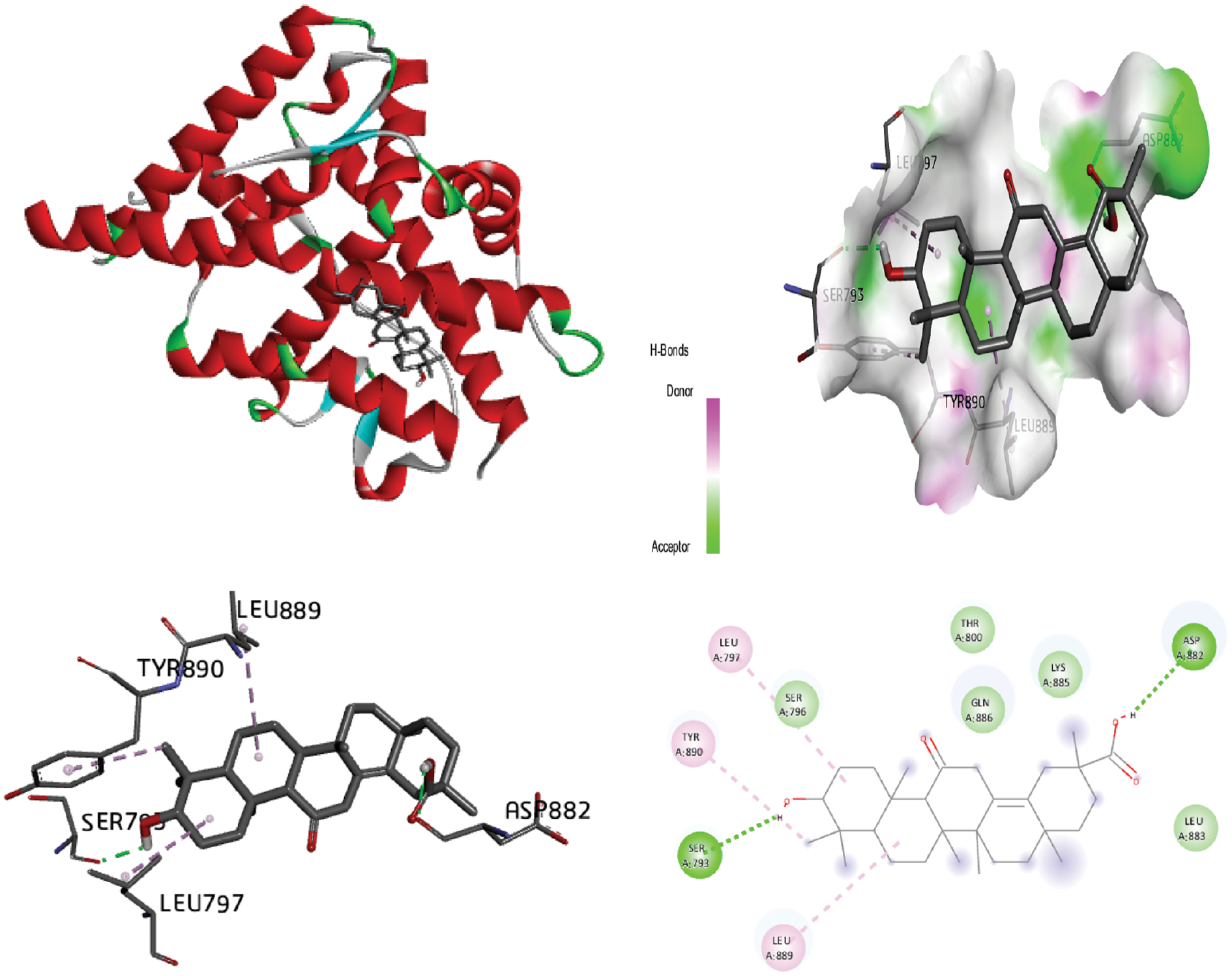
Figure 6 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with bisabola-3,10-dien-2-one shows 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
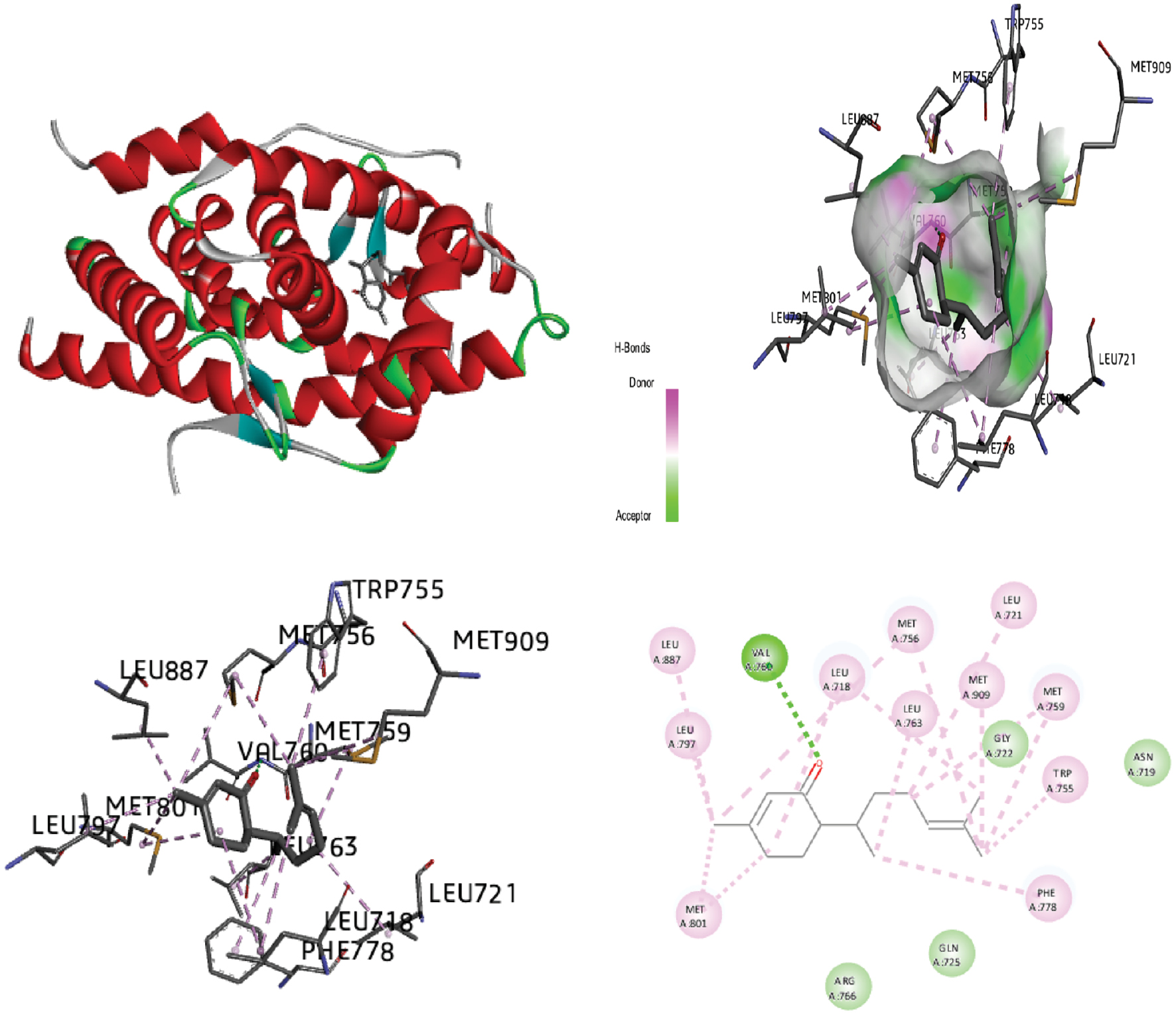
Figure 7 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with caffeic acid shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
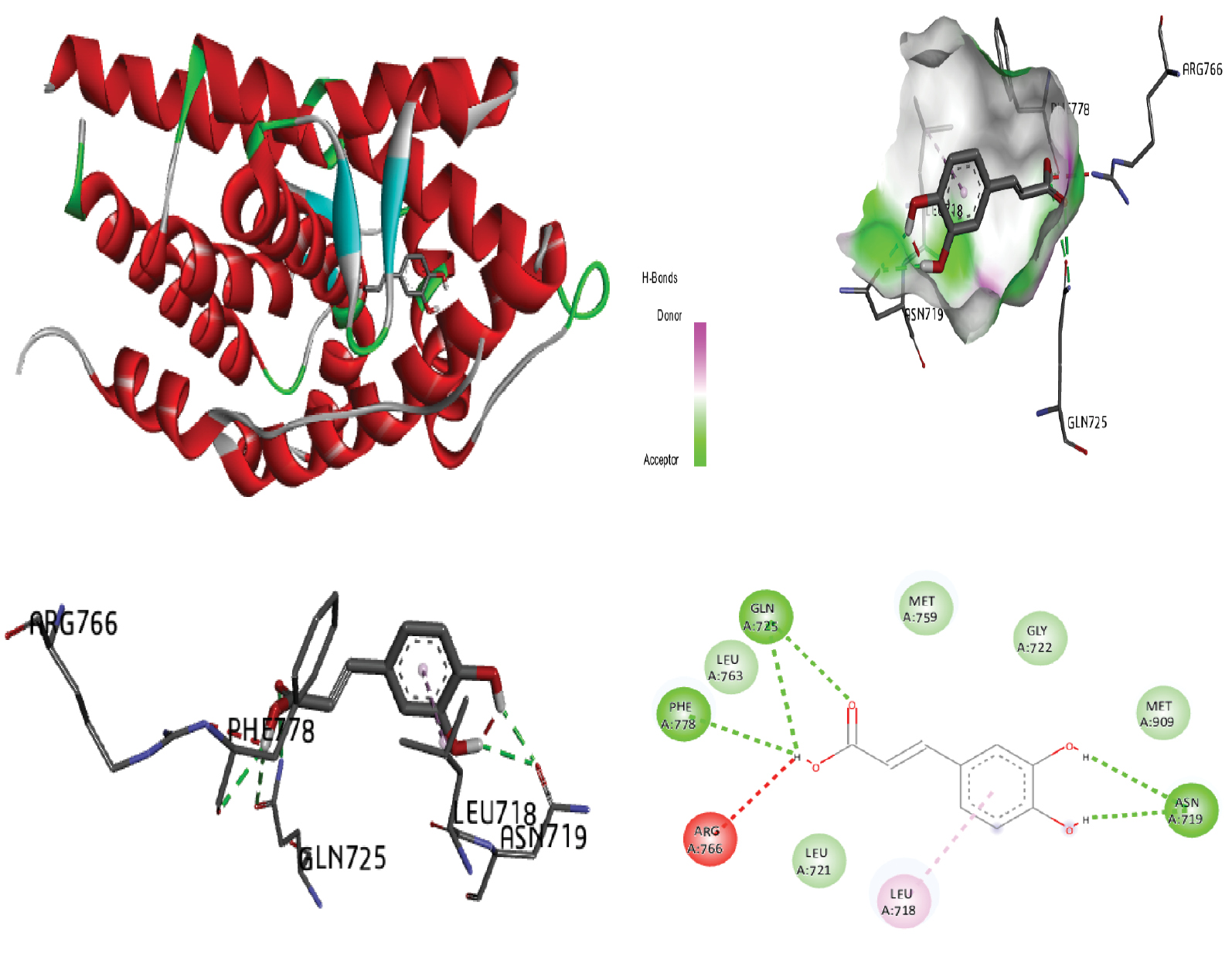
Figure 8 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with curdione shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
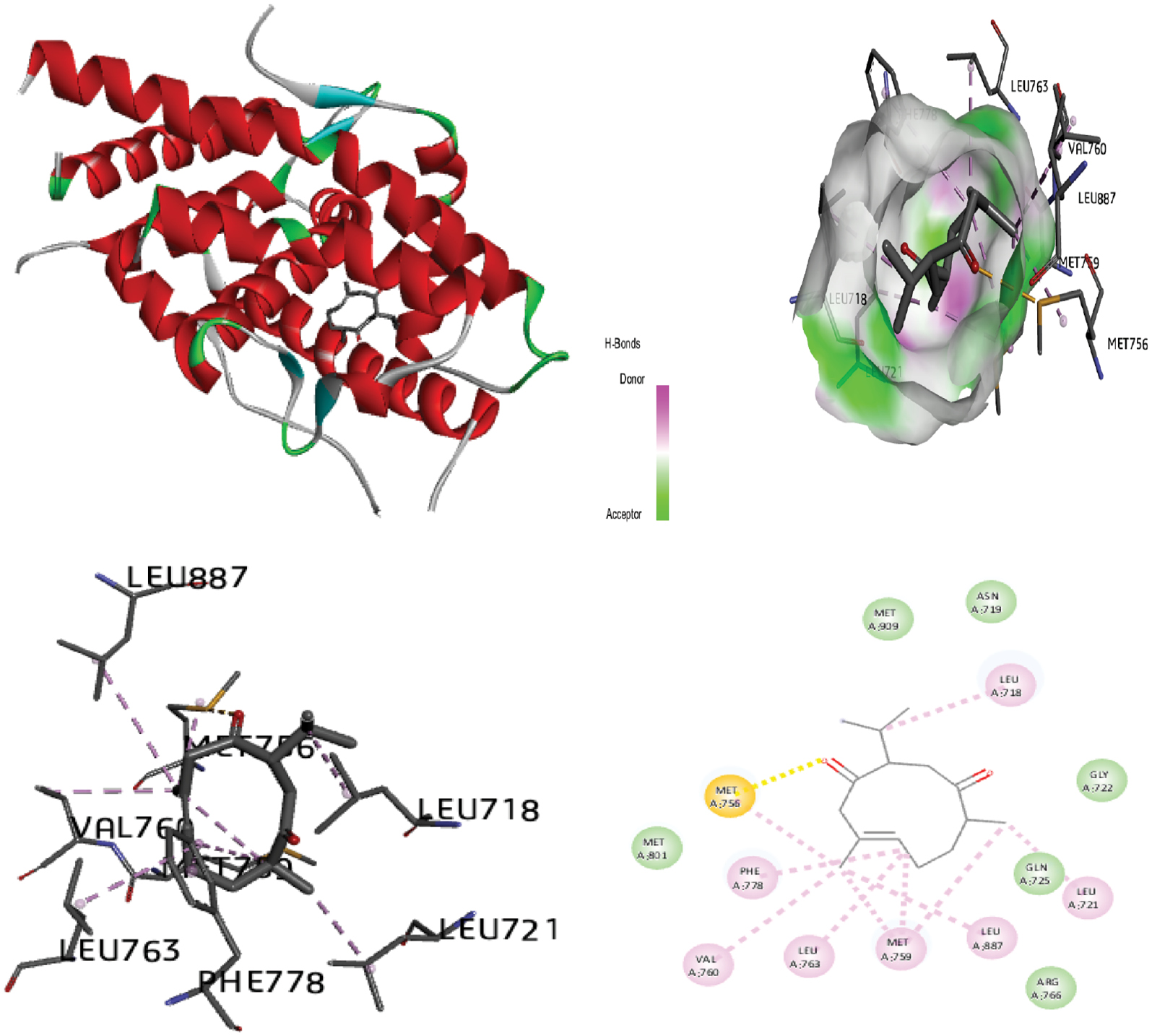
Figure 9 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with curcumenone shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
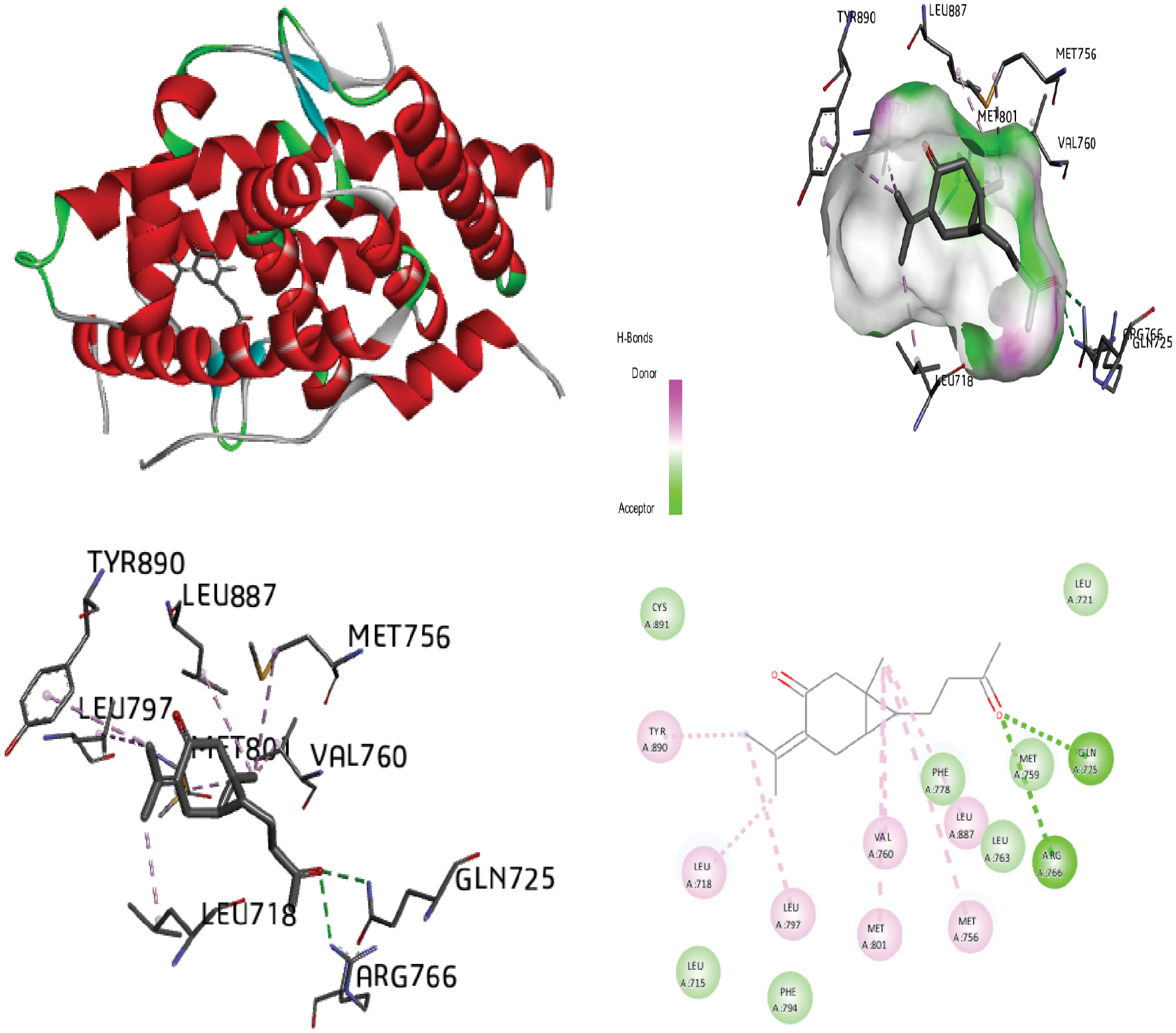
Figure 10 Molecular docking of the progesterone receptor (1SQN) binding domain complexed with dehydrocurdione shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
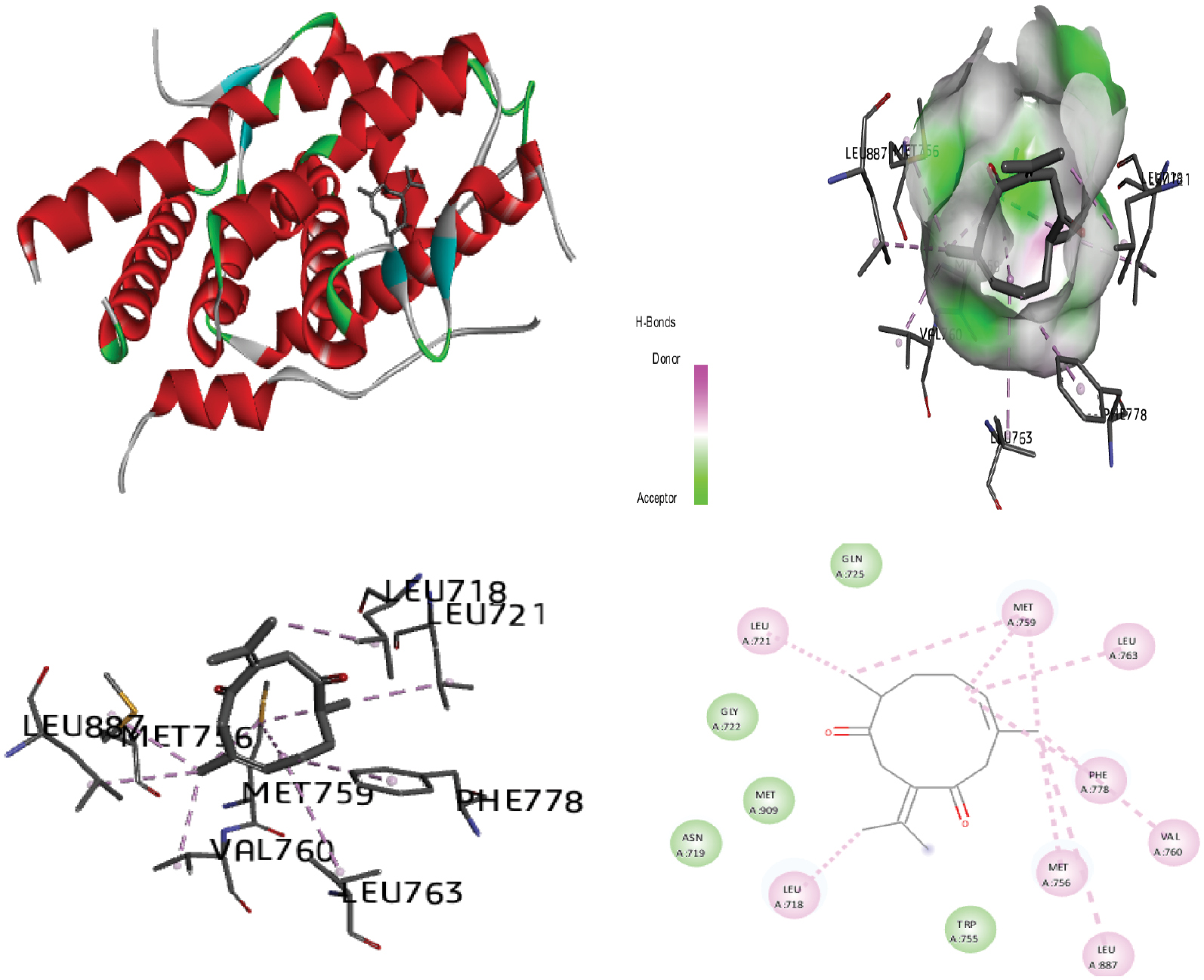
Figure 11 Molecular docking of the progesterone receptor (1SQN) binding domain Complexed with procurcumenol shows a 3D model of the interactions, and the 2D interaction patterns and H-bond interaction.
Table 1 PR Inhibition by Various Phytoconstituents
| Protein | Ligand | Binding Energy (kcal/mol) | No. H Bonds | Interacting Residue | Final Intermolecular Energy (kcal/mol) | vdW + Hbond + desolv Energy (kcal/mol) | Electrostatic Energy (kcal/mol) | Torsional Free Energy (kcal/mol) |
|---|---|---|---|---|---|---|---|---|
| 1SQN | Alpha-atlantone | −7.69 | 01 H1: 2.99 Å |
GLN:725 (H1) MET:759 LEU:721 LEU:718 PHE:778 MET:756 LEU:763 MET:909 VAL:760 MET:801 |
−8.28 | −8.18 | −0.10 | +0.89 |
| 18-Alpha-glycyrrhetinic acid | −6.36 | 03 H1: 1.87 Å H2: 2.98 Å H3: 2.76 Å |
ASP:882 (H1) GLN:886 (H2) ASN:893 (H3) LYS:885 TYR:890 |
−7.26 | −7.01 | −0.25 | +0.89 | |
| Alpha-turmerone | −7.85 | 01 H1: 3.07 Å |
GLN:725 (H1) VAL:760 MET:756 LEU:887 MET:759 LEU:797 TYR:890 MET:801 PHE:778 LEU:718 LEU:721 |
−8.61 | −8.52 | −0.09 | +1.19 | |
| 18-Beta-glycyrrhetinic acid | −5.38 | 02 H1: 2.09 Å H2: 2.08 Å |
SER:793 (H1) ASP:882 (H2) TYR:890 LEU:889 LEU:797 |
−6.24 | −6.36 | +0.12 | +0.89 | |
| Bisabola-3,10-dien-2-one | −7.56 | 01 H1: 3.32 Å |
VAL:760 (H1) LEU:887 LEU:797 MET:801 LEU:718 LEU:763 MET:756 MET:909 LEU:721 MET:759 TRP:755 PHE:778 |
−7.89 | −7.88 | −0.01 | +1.19 | |
| Caffeic acid | −5.87 | 03 H1: 2.59 Å H2: 2.87 Å H3: 2.16 Å |
PHE:778 (H1) GLN:725 (H2) ASN:719 (H3) ARG:766 LEU:718 |
−6.35 | −5.70 | −0.65 | +1.49 | |
| Curdione | −7.71 | 00 | LEU:718 LEU:721 LEU:887 MET:759 LEU:763 VAL:760 PHE:778 MET:756 |
−7.95 | −7.96 | +0.02 | +0.30 | |
| Curcumenone | −7.77 | 02 H1: 2.91 Å H2: 3.31 Å |
GLN:725 (H1) ARG:766 (H2) LEU:887 MET:756 VAL:760 MET:801 LEU:797 LEU:718 TYR:890 |
−8.73 | −8.61 | −0.12 | +0.89 | |
| Dehydrocurdione | −7.73 | 00 | LEU:721 LEU:718 MET:756 LEU:887 VAL:760 PHE:778 LEU:763 MET:759 |
−7.73 | −7.73 | +0.01 | +0.00 | |
| Procurcumenol | −7.85 | 01 H1: 2.17 Å |
LEU:718 (H1) LEU:721 MET:759 VAL:760 LEU:763 PHE:778 MET:801 |
−8.16 | −8.14 | −0.02 | +0.30 |
Lipinski’s rule of five was not violated by any compounds (Table 2). According to these guidelines, tested compounds were therefore anticipated to have good bioavailability and to satisfy drug likeliness characteristics [41].
Table 2 Lipinski’s Rule of Five and Other Information for Ligands
| No. | Name of Co-former | Molecular Formula | Mol. Weight (g/mol) | XlogP3 | Hydrogen Bond Donor | Hydrogen Bond Acceptor | Rotatable Bond |
|---|---|---|---|---|---|---|---|
| 1 | Alpha-atlantone | C15H10O5 | 218.33 | 4.1 | 0 | 1 | 3 |
| 2 | 18-Alpha-glycyrrhetinic acid | C28H48O | 470.7 | 6.4 | 2 | 4 | 1 |
| 3 | Alpha-turmerone | C27H42O3 | 218.33 | 3.8 | 0 | 1 | 4 |
| 4 | 18-Beta-glycyrrhetinic acid | C15H10O5 | 470.7 | 6.4 | 2 | 4 | 1 |
| 5 | Bisabola-3,10-dien-2-one | C16H12O5 | 220.35 | 4.1 | 0 | 1 | 4 |
| 6 | Caffeic acid | C15H10O7 | 180.16 | 1.2 | 3 | 4 | 2 |
| 7 | Curdione | C21H24O4 | 236.35 | 2.7 | 0 | 2 | 1 |
| 8 | Curcumenone | C29H50O | 234.33 | 2.4 | 0 | 2 | 3 |
| 9 | Dehydrocurdione | C28H42O5 | 234.33 | 2.8 | 0 | 2 | 0 |
| 10 | Procurcumenol | C17H14O8 | 234.33 | 2.3 | 1 | 2 | 0 |
CASTp identification
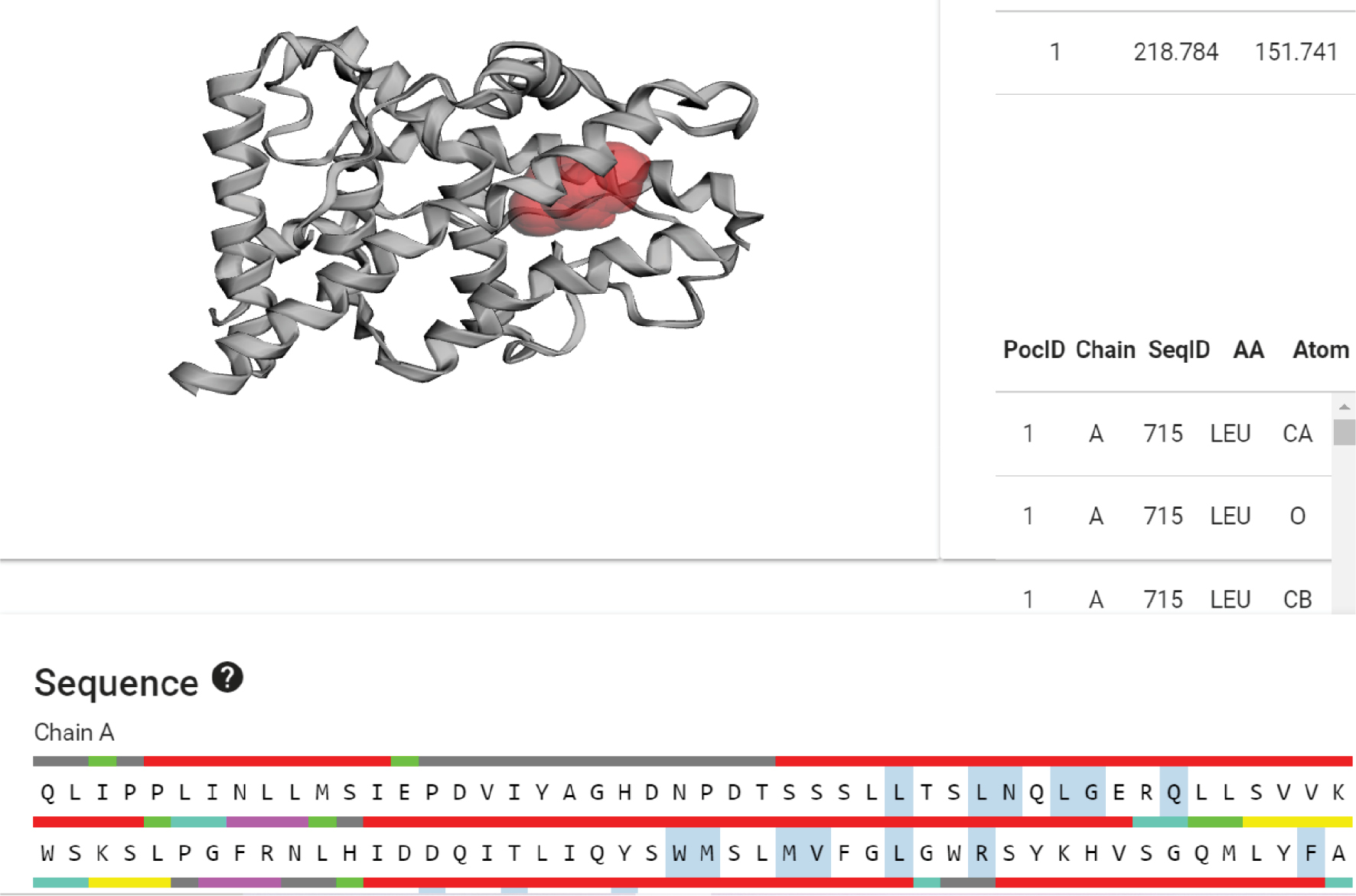
Based on the CASTp server analysis, we selected pocket 1, which had an area of 218.784 Å2 and a volume of 151.741 Å3. This pocket contained 22 amino acids (Figure 12) located at the following positions within the protein sequence: 715, 718, 719, 721, 722, 725, 755, 756, 759, 760, 763, 766, 778, 794, 797, 801, 887, 890, 891, 894, 905, and 909.
Figure 12 CASTp of PR.
ADMET studies
ADMET analysis, a computerized approach to drug development, can aid in early stages of drug discovery [41]. ADMET features aid in the analysis of new chemical substances to identify possible candidates that might be metabolized, cross membranes, and trigger or disable cellular functions [42].
A drug’s ability to cross membranes quickly and easily can be estimated by measuring or computing its octanol/water partitioning coefficient (logP) [43]. Alpha-atlantone, 18-alpha-glycyrrhetinic acid, and 18-beta-glycyrrhetinic acid exhibited logP values above 4. Additionally, the hydrophobic nature of bisabola-3,10-dien-2-one led to a logP value violation, with a value exceeding 5. The low logP value for caffeic acid was supported by the presence of hydrophilic hydroxyl groups. Occasionally, natural products’ lipophilicity does not precisely correlate with their physicochemical profiles, thus violating the rule of five [44]. Procurcumenol exhibited a lower logP (2.796) value than alpha-turmerone (3.956). For oral medications, a logP value of 2–3 is frequently considered ideal to balance permeation with first-pass clearance.
An essential characteristic of a drug-like molecule is solubility, given as log S, with optimal values ranging from −0.5 to −5.5 [45]. The solubility of all molecules was within the above-mentioned range, thereby indicating good solubility of each phytochemical. The highest solubility was observed for caffeic acid (−1.12), and the lowest solubility was observed for alpha-atlantone (−4.56). After oral administration, drugs are absorbed in the intestine and subsequently reach their specific targets. All molecules showed positive results regarding HIA. No molecules exhibited human intestinal permeability. Therefore, further work on permeability development is required for these drugs. Parameters such as BBB penetration and Caco-2 permeability have been studied to assess membrane permeability [46]. The BBB and Caco-2 values for all molecules showed positive values, thus suggesting an ability to easily cross barriers. Each tested molecule exhibited negative Caco-2 permeation values. All phytochemicals except alpha-atlantone, alpha-turmerone, and caffeic acid showed BBB permeability. The failure of BBB permeation by caffeic acid might have been due to its high hydrophilicity (logP 1.43). The absorption and permeability of drugs across the membranes such as the BBB can be optimized by using total polar surface area (TPSA) descriptors [47, 48]. When the TPSA exceeds 140 Å2, membrane permeation is normally restricted [49]. The TPSA values for all molecules ranged between 17.07 and 77.76.
P-glycoprotein (P-gp) efflux testing is a crucial step in the development of anti-cancer drugs, because P-gp is highly expressed in cancerous cells, where it prevents cells from absorbing chemotherapeutic drugs and causes cancerous cells to establish transporter-mediated resistance to anti-tumor therapies [50, 51]. P-gp substrates are transported back into the gastrointestinal lumen by P-gp, reducing the absorption and, consequently, the effectiveness of certain medications. The screened phytochemicals were found not to be P-gp substrates. All bioactives except 18-alpha-glycyrrhetinic acid, 18-beta-glycyrrhetinic acid, caffeic acid, dehydrocurdione, and procurcumenol were found to be P-gp inhibitors, thus enabling the dissemination of bioactives without restricting cellular absorption.
Furthermore, understanding drug binding to various plasma proteins is imperative, because of influences on drug therapeutic effects. The manner in which a drug attaches to plasma proteins crucially affects its physiological function [52]. All tested bioactives exhibited high PPB (>85%). The bioactives therefore would leave the body slowly and would require some time to start working, because only the free fraction of the chemical can initiate the desired therapeutic effect. Fraction unbound (Fu) is an important factor requiring precise quantification, because it markedly affects the calculation of therapeutic indices, the establishment of PK/PD correlations, and the prediction of interactions between drugs [53]. All chemicals showed a low Fu (<20%), which might potentially affect their clearance, pharmacological action, half-lives, and partitioning from blood to tissue. An essential drug-metabolizing enzyme in humans is CYP3A4. Cytochrome P450 (CYP450) is considered the primary parameter for assessing the ADME of medications, because of its function in phase I drug metabolism [54]. The inhibition of CYP3A4 can directly lead to drug toxicity, drug-drug interactions, and other adverse effects [41]. No tested phytochemicals were found to be inhibitors or substrates of CYP3A4.
LD50 values are frequently used for primary toxicity assessment, to describe the relative risks associated with the acute toxicity of substances. In general, more hazardous chemicals have lower LD50 values [49, 55]. All bioactives were found to be Ames test negative (non-toxic) and showed no signs of rat oral acute toxicity. However, most phytochemicals showed skin sensitization and carcinogenicity. All moieties except caffeic acid showed respiratory toxicity. Detailed results are described in Table 3.
Table 3 Performance of the Classification Models Incorporated Into the ADMETlab 2.0 Platform for Tested Bioactives
| Category | Model | Points | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
| Absorption | Caco-2 permeability | −4.42 | −5.42 | −4.56 | −5.42 | −4.50 | −5.22 | −4.58 | −4.58 | −4.53 | −4.60 |
| MDCK permeability | 2.3e-05 | 1.6e-05 | 1.9e-05 | 1.6e-05 | 1.6e-05 | 1.1e-05 | 1.8e-05 | 1.8e-05 | 2.1e-05 | 1.8e-05 | |
| Pgp inhibitor | + | −−− | ++ | −−− | + | −−− | ++ | ++ | −−− | −−− | |
| Pgp substrate | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | |
| HIA | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | |
| Distribution | PPB (%) | 97.83 | 93.09 | 93.81 | 93.09 | 96.41 | 87.71 | 88.85 | 88.85 | 85.30 | 82.38 |
| VD | 3.23 | 0.84 | 1.25 | 0.84 | 4.71 | 0.37 | 0.57 | 0.57 | 0.64 | 1.36 | |
| BBB penetration | −− | ++ | − | ++ | +++ | −− | +++ | +++ | +++ | + | |
| Fu (%) | 2.67 | 4.46 | 4.41 | 4.46 | 4.17 | 11.07 | 12.26 | 12.26 | 18.06 | 16.48 | |
| Metabolism | CYP1A2 inhibitor | ++ | −−− | −−− | −−− | + | −−− | −− | −− | −− | −−− |
| CYP1A2 substrate | +++ | + | ++ | + | − | −−− | ++ | ++ | − | −− | |
| CYP2C19 inhibitor | ++ | −−− | + | −−− | −− | −−− | + | + | − | −− | |
| CYP2C19 substrate | ++ | +++ | +++ | +++ | ++ | −−− | ++ | ++ | ++ | ++ | |
| CYP2C9 inhibitor | ++ | −− | − | −− | −− | −− | − | − | −− | −−− | |
| CYP2C9 substrate | + | − | + | − | +++ | −− | − | − | −− | −− | |
| CYP2D6 inhibitor | ++ | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | |
| CYP2D6 substrate | − | −− | ++ | −− | −− | −− | − | − | −− | −− | |
| CYP3A4 inhibitor | −−− | −− | − | −− | −− | −−− | −− | −− | −− | −−− | |
| CYP3A4 substrate | − | − | + | − | −− | −−− | − | − | − | − | |
| Excretion | CL | 7.58 | 2.53 | 14.31 | 2.53 | 17.85 | 10.97 | 13.38 | 13.38 | 14.11 | 14.35 |
| T1/2 | 0.66 | 0.07 | 0.80 | 0.07 | 0.35 | 0.93 | 0.85 | 0.85 | 0.87 | 0.52 | |
| Toxicity | hERG blockers | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− | −−− |
| H-HT | − | −− | ++ | −− | + | ++ | −− | −− | −−− | −−− | |
| DILI | −−− | −−− | −−− | −−− | − | − | − | − | + | −− | |
| Ames toxicity | −−− | −− | −−− | −− | −−− | −− | −− | −− | −− | −−− | |
| Rat oral acute toxicity | −−− | −− | −−− | −− | −−− | ++ | − | − | ++ | ++ | |
| FDAMDD | ++ | −− | +++ | −− | −−− | −−− | −− | −− | −−− | −− | |
| Skin sensitization | +++ | −−− | +++ | −−− | +++ | +++ | +++ | +++ | ++ | − | |
| Carcinogenicity | +++ | −−− | + | −−− | + | −− | ++ | ++ | ++ | ++ | |
| Respiratory toxicity | +++ | +++ | +++ | +++ | ++ | − | +++ | +++ | +++ | +++ | |
| BF | 2.35 | 0.46 | 1.28 | 0.46 | 0.84 | 0.44 | 0.49 | 0.49 | 0.56 | 0.52 | |
| IGC50 | 3.52 | 4.63 | 3.25 | 4.63 | 3.02 | 3.23 | 3.60 | 3.60 | 3.37 | 3.41 | |
| LC50FM | 4.32 | 5.45 | 4.35 | 5.45 | 4.45 | 3.75 | 4.53 | 4.53 | 4.71 | 4.32 | |
| LC50DM | 5.26 | 5.60 | 4.02 | 5.60 | 5.66 | 4.23 | 4.06 | 4.06 | 4.08 | 3.94 | |
| Physicochemical Properties | MW | 218.17 | 470.34 | 218.17 | 470.34 | 220.18 | 180.04 | 236.18 | 236.18 | 234.16 | 234.16 |
| Volume | 257.68 | 511.91 | 257.68 | 511.91 | 260.32 | 177.64 | 269.11 | 269.11 | 266.47 | 260.55 | |
| Density | 0.85 | 0.92 | 0.85 | 0.92 | 0.85 | 1.01 | 0.88 | 0.88 | 0.88 | 0.90 | |
| nHA | 1.00 | 4.00 | 1.00 | 4.00 | 1.00 | 4.00 | 2.00 | 2.00 | 2.00 | 2.00 | |
| nHD | 0.00 | 2.00 | 0.00 | 2.00 | 0.00 | 3.00 | 0.00 | 0.00 | 0.00 | 1.00 | |
| nRot | 3.00 | 1.00 | 4.00 | 1.00 | 4.00 | 2.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| nRing | 1.00 | 5.00 | 1.00 | 5.00 | 1.00 | 1.00 | 1.00 | 1.00 | 1.00 | 2.00 | |
| MaxRing | 6.00 | 22.00 | 6.00 | 22.00 | 6.00 | 6.00 | 10.00 | 10.00 | 10.00 | 10.00 | |
| nHet | 1.00 | 4.00 | 1.00 | 4.00 | 1.00 | 4.00 | 2.00 | 2.00 | 2.00 | 2.00 | |
| fChar | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | 0.00 | |
| nRig | 9.00 | 28.00 | 8.00 | 28.00 | 8.00 | 8.00 | 13.00 | 13.00 | 13.00 | 13.00 | |
| Flexibility | 0.33 | 0.04 | 0.50 | 0.04 | 0.50 | 0.25 | 0.00 | 0.00 | 0.00 | 0.00 | |
| Stereocenters | 1.00 | 8.00 | 2.00 | 8.00 | 2.00 | 0.00 | 2.00 | 2.00 | 2.00 | ||
| TPSA | 17.07 | 74.60 | 17.07 | 74.60 | 17.07 | 77.76 | 34.14 | 34.14 | 34.14 | 37.30 | |
| logS | −4.56 | −4.51 | −4.26 | −4.51 | −4.44 | −1.12 | −3.10 | −3.10 | −2.87 | −3.51 | |
| logP | 4.29 | 4.85 | 3.96 | 4.85 | 5.03 | 1.43 | 3.21 | 3.21 | 2.97 | 2.80 | |
| logD | 3.55 | 4.61 | 3.35 | 4.61 | 4.08 | 1.02 | 2.88 | 2.88 | 2.05 | 2.24 | |
The prediction probability values are indicated by six symbols: 0–0.1 (−−−), 0.1–0.3 (−−), 0.3–0.5 (−), 0.5–0.7 (+), 0.7–0.9 (++), and 0.9–1.0 (+++). 1: alpha-atlantone; 2: 18-alpha-glycyrrhetinic acid; 3: alpha-turmerone; 4: 18-beta-glycyrrhetinic acid; 5: bisabola-3,10-dien-2-one; 6: caffeic acid; 7: curdione; 8: curcumenone; 9: dehydrocurdione; 10: procurcumenol. MDCK: Madin-Darby canine kidney; Pgp: permeability glycoprotein; HIA: human intestinal absorption; PPB: plasma protein binding; VD: volume distribution; BBB: blood brain barrier; T1/2: half-life of the reaction; hERG: human ether à-go-go related gene; BF: bioconcentration factors; IGC50: 50% growth inhibitory concentration; MW: molecular weight; nHA: No. hydrogen acceptors; nHD: No. hydrogen donors; nRot: No. rotatable bonds; nRing: No. rings; MaxRing: maximum No. rings; nHet: No. heteroatoms; fChar: formal charge; nRig: No. rigid bonds; TPSA: topological polar surface area; logS: measuring solubility; logP: measuring lipophilicity; logD: distribution coefficient.
Conclusion
In this investigation, network analysis was instrumental in identifying plant bioactives capable of interacting with the PR. Moreover, the careful evaluation of ADMET parameters yielded critical insights into the drug likeliness, toxicity profiles, and permeability of these bioactives. Molecular docking further facilitated deeper exploration of their interaction mechanisms with the PR, and notably highlighted alpha-turmerone and procurcumenol as promising candidates for breast cancer therapeutics. These discoveries may provide a foundation for future endeavors aimed at formulating innovative dosage forms for breast cancer treatment. By enabling a nuanced understanding of the intricate interplay between bioactive constituents and the PR, this study may provide guidance for drug development and clinical translation. In the future, experimental validation will be imperative to corroborate these findings and enable practical implementation in breast cancer management.
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Gurung AB, Ali MA, Lee J, Farah MA, Al-Anazi KM. Molecular docking and dynamics simulation study of bioactive compounds from Ficus carica L. with important anticancer drug targets. PLoS One 2021;16(7):e0254035. [PMID: 34260631 DOI: 10.1371/journal.pone.0254035]
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, et al. Global Cancer Statistics 2020: GLOBOCAN Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J Clin 2021;71:209-49. [PMID: 33538338 DOI: 10.3322/caac.21660]
- Arnold M, Morgan E, Rumgay H, Mafra A, Singh D, et al. Current and future burden of breast cancer: global statistics for 2020 and 2040. Breast 2022;66:15-23. [PMID: 36084384 DOI: 10.1016/j.breast.2022.08.010]
- Konieczny M, Cipora E, Sygit K, Fal A. Quality of life of women with breast cancer and socio-demographic factors. Asian Pac J Cancer Prev 2020;21(1):185-93. [PMID: 31983183 DOI: 10.31557/APJCP.2020.21.1.185]
- Wilkinson L, Gathani T. Understanding breast cancer as a global health concern. Br J Radiol 2022;95(1130):20211033. [PMID: 34905391 DOI: 10.1259/bjr.20211033]
- Karpuz M, Silindir-Gunay M, Ozer AY. Current and future approaches for effective cancer imaging and treatment. Cancer Biother Radiopharm 2018;33(2):39-51. [PMID: 29634415 DOI: 10.1089/cbr.2017.2378]
- Liu YQ, Wang XL, He DH, Cheng YX. Protection against chemotherapy- and radiotherapy-induced side effects: a review based on the mechanisms and therapeutic opportunities of phytochemicals. Phytomedicine 2021;80:153402. [PMID: 33203590 DOI: 10.1016/j.phymed.2020.153402]
- Schirrmacher V. From chemotherapy to biological therapy: a review of novel concepts to reduce the side effects of systemic cancer treatment (Review). Int J Oncol 2019;54(2):407-19. [PMID: 30570109 DOI: 10.3892/ijo.2018.4661]
- Housman G, Byler S, Heerboth S, Lapinska K, Longacre M, et al. Drug resistance in cancer: an overview. Cancers (Basel) 2014;6(3):1769-92. [PMID: 25198391 DOI: 10.3390/cancers6031769]
- Durhan B, Yalçın E, Çavuşoğlu K, Acar A. Molecular docking assisted biological functions and phytochemical screening of Amaranthus lividus L. extract. Sci Rep 2022;12(1):4308. [PMID: 35279686 DOI: 10.1038/s41598-022-08421-8]
- Hashem S, Ali TA, Akhtar S, Nisar S, Sageena G, et al. Targeting cancer signaling pathways by natural products: exploring promising anti-cancer agents. Biomed Pharmacother 2022;150:113054. [PMID: 35658225 DOI: 10.1016/j.biopha.2022.113054]
- Deshpande SH, Muhsinah AB, Bagewadi ZK, Ankad GM, Mahnashi MH, et al. In silico study on the interactions, molecular docking, dynamics and simulation of potential compounds from Withania somnifera (L.) Dunal root against cancer by targeting KAT6A. Molecules 2023;28(3):1117. [PMID: 36770785 DOI: 10.3390/molecules28031117]
- Padma VV. An overview of targeted cancer therapy. Biomedicine (Taipei) 2015;5(4):19. [PMID: 26613930 DOI: 10.7603/s40681-015-0019-4]
- Antignani A, Ho ECH, Bilotta MT, Qiu R, Sarnvosky R, et al. Targeting receptors on cancer cells with protein toxins. Biomolecules 2020;10(9):1331. [PMID: 32957689 DOI: 10.3390/biom10091331]
- Hahn WC, Bader JS, Braun TP, Califano A, Clemons PA, et al. Cancer target discovery and development network. An expanded universe of cancer targets. Cell 2021;184(5):1142-55. [PMID: 33667368 DOI: 10.1016/j.cell.2021.02.020]
- Thakkar JP, Mehta DG. A review of an unfavorable subset of breast cancer: estrogen receptor positive progesterone receptor negative. Oncologist 2011;16(3):276-85. [PMID: 21339261 DOI: 10.1634/theoncologist.2010-0302]
- Pedroza DA, Subramani R, Tiula K, Do A, Rashiraj N, et al. Crosstalk between progesterone receptor membrane component 1 and estrogen receptor α promotes breast cancer cell proliferation. Lab Invest 2021;101(6):733-44. [PMID: 33903732 DOI: 10.1038/s41374-021-00594-6]
- Maennling AE, Tur MK, Niebert M, Klockenbring T, Zeppernick F, et al. Molecular targeting therapy against EGFR family in breast cancer: progress and future potentials. Cancers (Basel) 2019;11(12):1826. [PMID: 31756933 DOI: 10.3390/cancers11121826]
- Li Z, Wei H, Li S, Wu P, Mao X. The role of progesterone receptors in breast cancer. Drug Des Devel Ther 2022;16:305-14. [PMID: 35115765 DOI: 10.2147/DDDT.S336643]
- Bae SY, Kim S, Lee JH, Lee HC, Lee SK, et al. Poor prognosis of single hormone receptor-positive breast cancer: similar outcome as triple-negative breast cancer. BMC Cancer 2015;15:138. [PMID: 25880075 DOI: 10.1186/s12885-015-1121-4].
- Acharya R, Chacko S, Bose P, Lapenna A, Pattanayak SP. Structure based multitargeted molecular docking analysis of selected furanocoumarins against breast cancer. Sci Rep 2019;9(1):15743. [PMID: 31673107 DOI: 10.1038/s41598-019-52162-0]
- Mani S, Swargiary G, Gulati S, Gupta S, Jindal D. Molecular docking and ADMET studies to predict the anti-breast cancer effect of aloin by targeting estrogen and progesterone receptors. Mater Today Proc 2023;80(3):2378-84. [DOI: 10.1016/j.matpr.2021.06.362]
- Ismail S, Uzairu A, Sagagi B, Suleiman MS. Insilico molecular docking and pharmacokinetic studies of selected phytochemicals with estrogen and progesterone receptors as anticancer agent for breast cancer. J Turkish Chem Soc A Chem 2018;5(3):1337-50. [DOI: 10.18596/jotcsa.449778]
- Gnanaselvan S, Yadav SA, Manoharan SP. Structure-based virtual screening of anti-breast cancer compounds from Artemisia absinthium-insights through molecular docking, pharmacokinetics, and molecular dynamic simulations. J Biomol Struct Dyn 2024;42(6):3267-85. [PMID: 37194295 DOI: 10.1080/07391102.2023.2212805]
- Maia EHB, Assis LC, de Oliveira TA, da Silva AM, Taranto AG. Structure-based virtual screening: from classical to artificial intelligence. Front Chem 2020;8:343. [PMID: 32411671 DOI: 10.3389/fchem.2020.00343]
- Gimeno A, Ojeda-Montes MJ, Tomás-Hernández S, Cereto-Massagué A, Beltrán-Debón R, et al. The light and dark sides of virtual screening: what is there to know? Int J Mol Sci 2019;20(6):1375. [PMID: 30893780 DOI: 10.3390/ijms20061375]
- Ralte L, Khiangte L, Thangjam NM, Kumar A, Singh YT. GC-MS and molecular docking analyses of phytochemicals from the underutilized plant, Parkia timoriana revealed candidate anti-cancerous and anti-inflammatory agents. Sci Rep 2022;12(1):3395. [PMID: 35233058 DOI: 10.1038/s41598-022-07320-2]
- Taghizadeh MS, Niazi A, Moghadam A, Afsharifar A. Experimental, molecular docking and molecular dynamic studies of natural products targeting overexpressed receptors in breast cancer. PLoS One 2022;17(5):e0267961. [PMID: 35536789 DOI: 10.1371/journal.pone.0267961]
- Chandran U, Patwardhan B. Network ethnopharmacological evaluation of the immunomodulatory activity of Withania somnifera. J Ethnopharmacol 2017;197:250-6. [PMID: 27487266 DOI: 10.1016/j.jep.2016.07.080]
- U.S. Department of Agriculture, Agricultural Research Service. 1992-2016. Dr. Duke’s phytochemical and ethnobotanical databases. Available from: http://phytochem.nal.usda.gov/ (Home Page). [DOI: 10.15482/USDA.ADC/1239279]
- Vivek-Ananth RP, Mohanraj K, Sahoo AK, Samal A. IMPPAT 2.0: an enhanced and expanded phytochemical atlas of Indian medicinal plants. ACS Omega 2023;8(9):8827-45. [PMID: 36910986 DOI: 10.1021/acsomega.3c00156]
- Kim S, Chen J, Cheng T, Gindulyte A, He J, et al. PubChem 2023 update. Nucleic Acids Res 2023;51(D1):D1373-80. [PMID: 36305812 DOI: 10.1093/nar/gkac956]
- Berman HM, Bhat TN, Bourne PE, Feng Z, Gilliland G, et al. The Protein Data Bank and the challenge of structural genomics. Nat Struct Biol 2000;7 Suppl:957-9. [PMID: 11103999 DOI: 10.1038/80734]
- Integrative Biomedical Informatics Group GRIB/IMIM/UPF. Gene-disease association data retrieved from DisGeNET v7.0. Available from: https://www.disgenet.org/.
- UniProt Consortium. UniProt: the universal protein knowledgebase in 2023. Nucleic Acids Res 2023;51(D1):D523-31. [PMID: 36408920 DOI: 10.1093/nar/gkac1052]
- Ko M, Kim Y, Kim HH, Jeong S, Ahn D, et al. Network pharmacology and molecular docking approaches to elucidate the potential compounds and targets of Saeng-Ji-Hwang-Ko for treatment of type 2 diabetes mellitus. Comput Biol Med 2022;149:106041. [PMID: 36049411 DOI: 10.1016/j.compbiomed.2022.106041]
- Li S, Zhang B. Traditional Chinese medicine network pharmacology: theory, methodology and application. Chin J Nat Med 2013;11:110-20. [PMID: 23787177 DOI: 10.1016/S1875-5364(13)60037-0]
- Bourzikat O, El Abbouchi A, Ghammaz H, El Brahmi N, El Fahime E, et al. Synthesis, anticancer activities and molecular docking studies of a novel class of 2-phenyl-5,6,7,8-tetrahydroimidazo [1,2-b]pyridazine derivatives bearing sulfonamides. Molecules 2022;27(16):5238. [PMID: 36014478 DOI: 10.3390/molecules27165238]
- Deodware SA, Barache UB, Dhale PC, Gaikwad KD, Shivamallu C, et al. In vitro anticancer screening, molecular docking and antimicrobial studies of triazole-based nickel(II) metal complexes. Molecules 2022;27(19):6548. [PMID: 36235085 DOI: 10.3390/molecules27196548]
- Dundas J, Ouyang Z, Tseng J, Binkowski A, Turpaz Y, et al. CASTp: computed atlas of surface topography of proteins with structural and topographical mapping of functionally annotated residues. Nucleic Acids Res 2006;34(Web Server issue):W116-8. [PMID: 16844972 DOI: 10.1093/nar/gkl282]
- Awadelkareem AM, Al-Shammari E, Elkhalifa AEO, Adnan M, Siddiqui AJ, et al. Phytochemical and in silico ADME/Tox analysis of Eruca sativa extract with antioxidant, antibacterial and anticancer potential against Caco-2 and HCT-116 colorectal carcinoma cell lines. Molecules 2022;27(4):1409. [PMID: 35209197 DOI: 10.3390/molecules27041409]
- Gouthami K, Veeraraghavan V, Nagaraja P. In-silico characterization of phytochemicals identified from Vitex negundo (L) extract as potential therapy for Wnt-signaling proteins. Egypt J Med Hum Genet 2022;23(3):1-15. [DOI: 10.1186/s43042-022-00219-7]
- Carpenter TS, Kirshner DA, Lau EY, Wong SE, Nilmeier JP, et al. A method to predict blood-brain barrier permeability of drug-like compounds using molecular dynamics simulations. Biophys J 2014;107(3):630-41. [PMID: 25099802 DOI: 10.1016/j.bpj.2014.06.024]
- Raju L, Lipin R, Eswaran R. Identification, ADMET evaluation and molecular docking analysis of Phytosterols from Banaba (Lagerstroemia speciosa (L.)Pers) seed extract against breast cancer. In Silico Pharmacol 2021;9(1):43. [PMID: 34367875 DOI: 10.1007/s40203-021-00104-y]
- Joshi T, Joshi T, Sharma P, Pundir H, Chandra S. In silico identification of natural fungicide from Melia azedarach against isocitrate lyase of Fusarium graminearum. J Biomol Struct Dyn 2021;39(13):4816-34. [PMID: 32568603 DOI: 10.1080/07391102.2020.1780941]
- Niveshika, Singh S, Verma E, Mishra AK. In silico molecular docking analysis of cancer biomarkers with GC/MS identified compounds of Scytonema sp. Netw Model Anal Health Inform Bioinform 2020;9:30. [DOI: 10.1007/s13721-020-00235-w]
- Shityakov S, Neuhaus W, Dandekar T, Förster C. Analysing molecular polar surface descriptors to predict blood-brain barrier permeation. Int J Comput Biol Drug Des 2013;6(1-2):146-56. [PMID: 23428480 DOI: 10.1504/IJCBDD.2013.052195]
- Tan NC, Yu P, Kwon YU, Kodadek T. High-throughput evaluation of relative cell permeability between peptoids and peptides. Bioorg Med Chem 2008;16(11):5853-61. [PMID: 18490170 DOI: 10.1016/j.bmc.2008.04.074]
- Matsson P, Kihlberg J. How big is too big for cell permeability? J Med Chem 2017;60(5):1662-4. [PMID: 28234469 DOI: 10.1021/acs.jmedchem.7b00237]
- Amin ML. P-glycoprotein inhibition for optimal drug delivery. Drug Target Insights 2013;7:27-34. [PMID: 24023511 DOI: 10.4137/DTI.S12519]
- Li N, Kulkarni P, Badrinarayanan A, Kefelegn A, Manoukian R, et al. P-glycoprotein substrate assessment in drug discovery: application of modeling to bridge differential protein expression across in vitro tools. J Pharm Sci 2021;110(1):325-37. [PMID: 32946896 DOI: 10.1016/j.xphs.2020.09.017]
- Keen P. Effect of binding to plasma proteins on the distribution, activity and elimination of drugs. In: Brodie BB, Gillette JR, Ackerman HS, editors. Concepts in biochemical pharmacology. Handbook of experimental pharmacology, Vol. 28/1. Berlin, Heidelberg: Springer; 1971. [DOI: 10.1007/978-3-642-65052-9_10]
- Di L. An update on the importance of plasma protein binding in drug discovery and development. Expert Opin Drug Discov 2021;16(12):1453-65. [PMID: 34403271 DOI: 10.1080/17460441.2021.1961741]
- Paramashivam SK, Elayaperumal K, Natarajan BB, Ramamoorthy MD, Balasubramanian S, et al. In silico pharmacokinetic and molecular docking studies of small molecules derived from Indigofera aspalathoides Vahl targeting receptor tyrosine kinases. Bioinformation 2015;11(2):73-84. [PMID: 25848167 DOI: 10.6026/97320630011073]
- Ruiz P, Begluitti G, Tincher T, Wheeler J, Mumtaz M. Prediction of acute mammalian toxicity using QSAR methods: a case study of sulfur mustard and its breakdown products. Molecules 2012;17(8):8982-9001. [PMID: 22842643 DOI: 10.3390/molecules17088982]