Lactic acid and Lactylation in the Progression of Pulmonary Fibrosis
1Institute for Applied Research in Public Health, Nantong Key Laboratory of Environmental Toxicology, School of Public Health, Nantong University, Nantong 226019, China
2Division of Oral and Systemic Health Sciences, School of Dentistry, University of California, Los Angeles, CA 90095, USA
aThese authors contributed equally to this work.
*Correspondence to: Bing Han, Division of Oral and Systemic Health Sciences, School of Dentistry, University of California, Los Angeles, Los Angeles, CA 90095, USA, E-mail: binghan@ucla.edu; Demin Cheng, Institute for Applied Research in Public Health, Nantong Key Laboratory of Environmental Toxicology, School of Public Health, Nantong University, Nantong 226019, China, E-mail: cdm95101@126.com;Xinyuan Zhao, Institute for Applied Research in Public Health, Nantong Key Laboratory of Environmental Toxicology, School of Public Health, Nantong University, Nantong 226019, China, E-mail: zhaoxinyuan@ntu.edu.cn
Received: January 13 2026; Revised: April 18 2026; Accepted: May 3 2026; Published Online: May 26 2026
Cite this paper:
Wang F, Jiang M, Zhu L et al. Lactic acid and Lactylation in the Progression of Pulmonary Fibrosis. BIO Integration 2026; 7: 1–34.
DOI: 10.15212/bioi-2026-0009. Available at: https://bio-integration.org/
Download citation
© 2026 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Pulmonary fibrosis (PF) is a progressive and irreversible interstitial lung disease that is characterized by destruction of alveolar architecture, excessive proliferation of fibroblasts, and aberrant deposition of extracellular matrix (ECM) but the precise pathogenesis has yet to be fully elucidated. Although traditionally regarded as the terminal metabolite of glycolysis, lactate has been reappreciated, especially following the rise of metabolic reprogramming concepts after the Warburg effect, as an important signaling molecule capable of actively regulating diverse cellular functions. Among these cellular functions, the lactate-induced post-translational modification (PTM), known as lactylation, offers a new perspective for understanding the broad biological actions of lactate beyond metabolism. Notably, the pathologic microenvironment of PF is characterized by widespread metabolic reprogramming and lactate accumulation, suggesting that lactate and lactate-mediated lactylation may serve key roles in disease progression by regulating pro-fibrotic gene expression and influencing fibroblast activation and differentiation. Therefore, this review focuses on how lactylation functions as a bridge linking metabolic reprogramming to fibrotic phenotypes in PF and the translational potential as a novel therapeutic target is discussed.
Keywords
Eraser, lactate shuttle, lactic acid, lactylation, metabolic reprogramming, pulmonary fibrosis, reader, writer.
Introduction
Pulmonary fibrosis (PF) is a chronic, progressive lung disease that is characterized by fibroblast proliferation and widespread deposition of the extracellular matrix (ECM) and collagen, accompanied by inflammatory injury, and ultimately leading to death from respiratory failure [1, 2]. The pathogenesis underlying PF is highly complex and widely believed to be driven not by a single factor but by a multicellular network process. Repetitive injury to alveolar epithelial cells is the key inciting event, triggering persistent activation and interactions among multiple cell types, including immune and mesenchymal cells, which together promote malignant progression of fibrosis [3–5].
Within this dynamic cellular network, diverse cells orchestrate disease progression through complex signaling dialogues [3]. Damaged type II alveolar epithelial cells (AEC2s) not only secrete pro-fibrotic factors, such as transforming growth factor-beta (TGF-β) and platelet-derived growth factor (PDGF), but also participate in the formation of fibrotic phenotypes through epithelial–mesenchymal transition (EMT)–related processes [4, 6]. These signals recruit and aberrantly activate pulmonary fibroblasts, driving differentiation into highly secretory myofibroblasts that become the major source of excessive ECM production [7]. Infiltration and polarization of immune cells (e.g., macrophages, neutrophils, and lymphocytes) act as “amplifiers” within this network [8]. Among the amplifiers, M2 macrophages are regarded as key pro-fibrotic immune cells that further enhance fibroblast activation and proliferation by secreting pro-fibrotic cytokines and growth factors [9].
In recent years metabolic reprogramming has been confirmed as a central driver of this pathogenic cellular network [10]. The fibrotic microenvironment in PF features hypoxia and abnormal energy metabolism. Notably, multiple core cell types, including activated fibroblasts, M2 macrophages, and dysfunctional epithelial cells, exhibit markedly enhanced glycolysis. An inevitable consequence of glycolysis is the production and abundant release of lactate, resulting in significantly elevated local lactate levels in PF lesions [11]. Once considered a metabolic waste, lactate has now been shown to have a crucial signaling role in fibrosis [12]. A high lactate level not only directly stimulates collagen synthesis in fibroblasts and promotes macrophage polarization toward the M2 phenotype but also suppresses immune cell function, thereby forming a pro-fibrotic positive-feedback loop [13, 14].
Lactate has emerged as a molecule of growing interest in disease biology as a metabolic intermediate and a direct regulator of gene expression through epigenetic mechanisms [15, 16]. The identification of histone lactylation in 2019 as a novel lysine post-translational modification marked a conceptual shift in our understanding of lactate from a metabolic byproduct to an active epigenetic regulator. Lactylation refers to the covalent addition of lactyl groups derived from lactate to lysine residues on histone and non-histone proteins, thereby modulating chromatin architecture, transcriptional activity, and protein function [17–19]. Among these modifications, H3K18la has been most extensively characterized and is closely associated with transcriptional activation of specific gene programs [17]. These findings suggested that lactate accumulation is not merely a metabolic signature of pathologic states but can be inscribed into stable epigenetic marks, effectively converting transient metabolic perturbations into sustained transcriptional reprogramming.
Lactylation represents a mechanistic interface between cellular metabolism and epigenetic regulation. Lactylation is inherently dependent on intracellular lactate availability and acyl-donor pools, linking lactylation tightly to glycolytic flux, redox homeostasis, and microenvironmental metabolic stress. Conversely, the dynamic regulation of lactylation likely involves dedicated enzymatic systems, including acetyltransferase-like “writers” and deacylase “erasers” [20–22]. Although the full repertoire of lactylation regulators, including specific writers, erasers, and readers, remains incompletely defined, accumulating evidence indicates that lactylation is a regulated and functionally selective process rather than a passive chemical consequence [18, 23]. Notably, in contrast to classical epigenetic modifications, such as acetylation and methylation, lactylation directly encodes metabolic information, thereby providing a conceptual framework through which metabolic dysregulation can be translated into stable transcriptional programs.
This framework is particularly pertinent in PF, a disease that is characterized by persistent metabolic reprogramming and a chronically lactate-enriched microenvironment. The sustained elevation of lactate in fibrotic lesions provides a biochemical substrate for lactylation, while key cellular players, including injured epithelial cells, activated fibroblasts, and polarized macrophages, exhibit heightened glycolytic activity and marked phenotypic plasticity. Within this context, lactate is likely to function as a signaling metabolite and an epigenetic substrate that drives histone and non-histone lactylation, thereby fine-tuning core fibrotic processes, such as TGF-β signaling, extracellular matrix production, inflammatory amplification, and cell fate transitions [24]. Thus, lactylation may serve as a critical molecular node linking metabolic rewiring to transcriptional reprogramming and ultimately to the stabilization of pro-fibrotic phenotypes.
Despite the emerging importance of lactylation, the role of lactylation in PF is incompletely understood. Current studies are largely fragmented and tend to focus on lactate accumulation or metabolic alterations in isolation with limited integration of lactylation-dependent mechanisms. Specifically, how metabolic imbalance is translated into coordinated intercellular communication through lactylation and how this process amplifies fibrotic progression is poorly defined. Key questions regarding the molecular circuitry of lactylation, the cell-type-specific functions, and the translational potential in PF have not been resolved. This review aims to address these gaps by providing a systematic and integrative perspective on lactylation in PF. We propose that lactylation functions as a central axis linking metabolic reprogramming to fibrotic gene expression and further highlight the cell-type-specific roles of lactylation and potential as a therapeutic target, thereby offering a refined conceptual framework for understanding the pathogenesis underlying fibrosis.
Lactate
Sources of lactate
Lactate is a key three-carbon metabolic intermediate with broad origins that span biological activities from microbial fermentation to cellular metabolism in higher organisms [25]. The classic source of lactate is cellular glycolysis. Under hypoxic conditions or when energy demand surges, such as in exercising skeletal muscle or rapidly proliferating tumor cells, glucose is converted to pyruvate through glycolysis, then reduced to lactate by lactate dehydrogenase A (LDHA), concomitantly regenerating nicotinamide adenine dinucleotide (NAD+) to sustain a high glycolytic flux [26, 27]. Figure 1 provides a detailed explanation of the sources of lactic acid.
Figure 1 Multiple sources of lactic acid. Lactic acid primarily originates from cellular glycolysis. Under hypoxic conditions or when energy demand surges (e.g., in skeletal muscle during exercise or proliferating tumor cells), glucose undergoes glycolysis to produce pyruvate, which is subsequently reduced to lactic acid by lactate dehydrogenase A, while simultaneously regenerating NAD+ to maintain high glycolytic flux. Tumor cells exhibit high dependence on glycolysis and substantial lactic acid production even under aerobic conditions, a phenomenon termed aerobic glycolysis or Warburg effect, which serves as a hallmark of cancer metabolic reprogramming and is regulated by pathways such as PI3K/Akt/mTOR. Upregulation of pyruvate dehydrogenase kinase 4 inhibits pyruvate entry into mitochondria in senescent cells, similarly promoting aerobic glycolysis and lactic acid production. Studies have confirmed that the sustained generation and utilization of lactic acid in various aerobic cells act as a critical nexus linking glycolysis and oxidative metabolism. Lactic acid participates in inter-organ metabolic coordination through the Cori cycle at the systemic level. Lactic acid produced by skeletal muscle is taken up by the liver via the bloodstream and converted into glucose through gluconeogenesis to maintain blood glucose homeostasis and energy balance, a process validated by techniques, such as isotope tracing. Thus, lactic acid is not only a metabolic product but also an important energy carrier and signaling molecule.
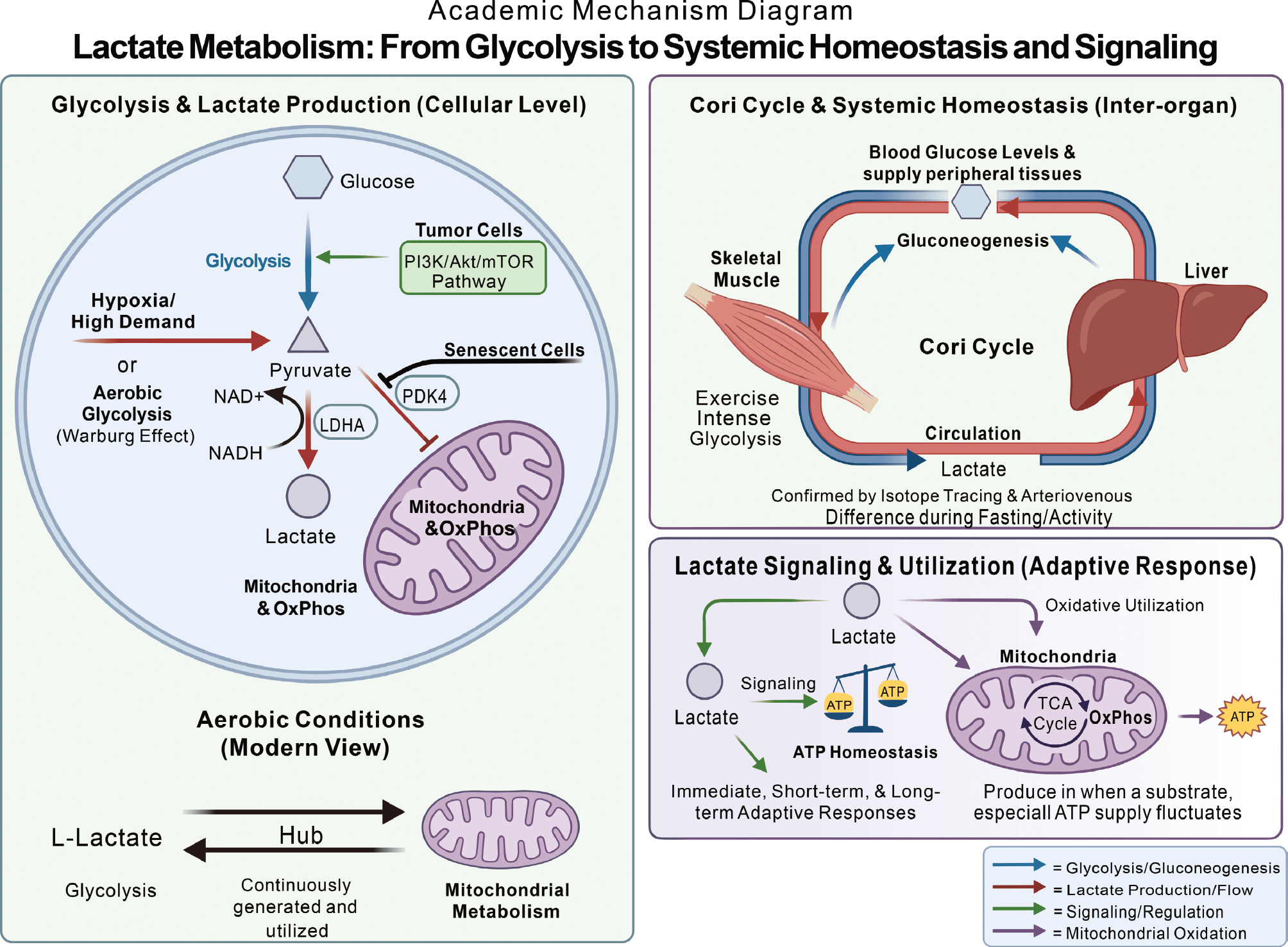
This metabolic mode, high dependence on glycolysis and abundant lactate production even under aerobic conditions, is termed aerobic glycolysis or the Warburg effect in tumor cells, a hallmark of cancer metabolic reprogramming that supplies energy and multiple biosynthetic precursors for rapid growth [28–31]. Studies have shown that activation of the PI3K/Akt/mTOR pathway markedly enhances glycolytic throughput and leads to excessive lactate generation in malignancies, such as leukemia [32, 33]. Upregulation of pyruvate dehydrogenase kinase 4 (PDK4) in senescent cells inhibits mitochondrial entry of pyruvate, similarly boosting aerobic glycolysis and lactate production, a highly catabolic state closely associated with the senescence-associated secretory phenotype and age-related pathology [34].
Contrary to the traditional view that lactate arises mainly from inadequate oxygen supply during skeletal muscle contraction, extensive research has demonstrated that the L-enantiomer of lactate is continuously generated and utilized in many cell types even under fully aerobic conditions [35]. As the terminal product of glycolysis and a downstream substrate for mitochondrial aerobic metabolism, lactate can be considered an important hub connecting glycolysis and oxidative pathways [35]. Aerobic respiration is jointly executed by cytosolic and mitochondrial compartments under physiologic conditions. Although initially labeled a metabolic waste and fatigue factor, lactate serves as a key signaling molecule within the complex metabolic feedback network. When ATP supply fluctuates transiently, lactate production rapidly initiates immediate, short-term, and long-term adaptive responses that help maintain ATP homeostasis [36].
Lactate metabolism mainly relies on oxidative utilization. Thus, earlier studies focused on the role of lactate as an oxidative substrate. From the perspective of “beneficial metabolism,” the earliest discovery revealing the advantage of lactate metabolism is the Cori cycle, a classic model in which lactate serves as the principal precursor for gluconeogenesis to maintain systemic energy homeostasis. Lactate shuttling in the Cori cycle interconnects intra- and inter-organ networks (muscle to liver). Specifically, skeletal muscle releases lactate into the circulation when glycolysis intensifies, then the liver takes up lactate and converts lactate back to glucose, which is then redistributed to peripheral tissues. It has been confirmed using modern isotope tracing and arteriovenous difference measurements that under various physiologic states, including fasting at rest and physical activity, lactate contributes substantially to gluconeogenesis and helps maintain relatively stable blood glucose levels [37].
Core lactate catabolic pathways
Lactate degradation mainly proceeds through two core routes: (1) reconversion to pyruvate followed by entry into the tricarboxylic acid (TCA) cycle for complete oxidative energy production; and (2) conversion to lactyl-CoA as the direct donor for protein lactylation, thereby transducing metabolic signals into regulation of protein function and gene expression [17, 26]. These two pathways constitute the central routes of lactate catabolism, coupling energy metabolism with epigenetic and signaling regulation, as illustrated in Figure 2.
Figure 2 Dual core pathways of lactic acid catabolism: Energy production and protein lactylation signaling. This figure elucidates the two core metabolic pathways of lactic acid catabolism. Pathway 1 (Energy Production): Lactic acid is reversibly oxidized to pyruvate by lactate dehydrogenase (LDH), which then enters the mitochondria. Under the action of the pyruvate dehydrogenase complex, pyruvate is converted to acetyl-CoA, which subsequently enters the tricarboxylic acid (TCA) cycle and efficiently produces ATP through oxidative phosphorylation. Studies have shown that the lactylation of the PDC subunit E1α (PDHA1) can inhibit the activity under hypoxic conditions, thereby precisely regulating metabolic flux. Pathway 2 (Signaling/Lactylation): Lactic acid is converted into the high-energy intermediate, lactoyl-CoA, which is covalently modified onto the lysine residues of target proteins by specific “writer” enzymes, resulting in protein lactylation. Lactoyl-CoA can originate from intracellular lactic acid produced by glycolysis or be derived from extracellular lactic acid transported by monocarboxylate transporters. Lactylation modifications also include non-enzymatic mechanisms independent of lactoyl-CoA and reversible processes mediated by histone deacetylases of class I, thereby translating fluctuations in intracellular lactic acid levels into regulation of protein function and gene expression (e.g., epigenetic regulation). These two pathways collectively form the core framework of lactic acid catabolism, tightly coupling energy metabolism with cellular signaling.
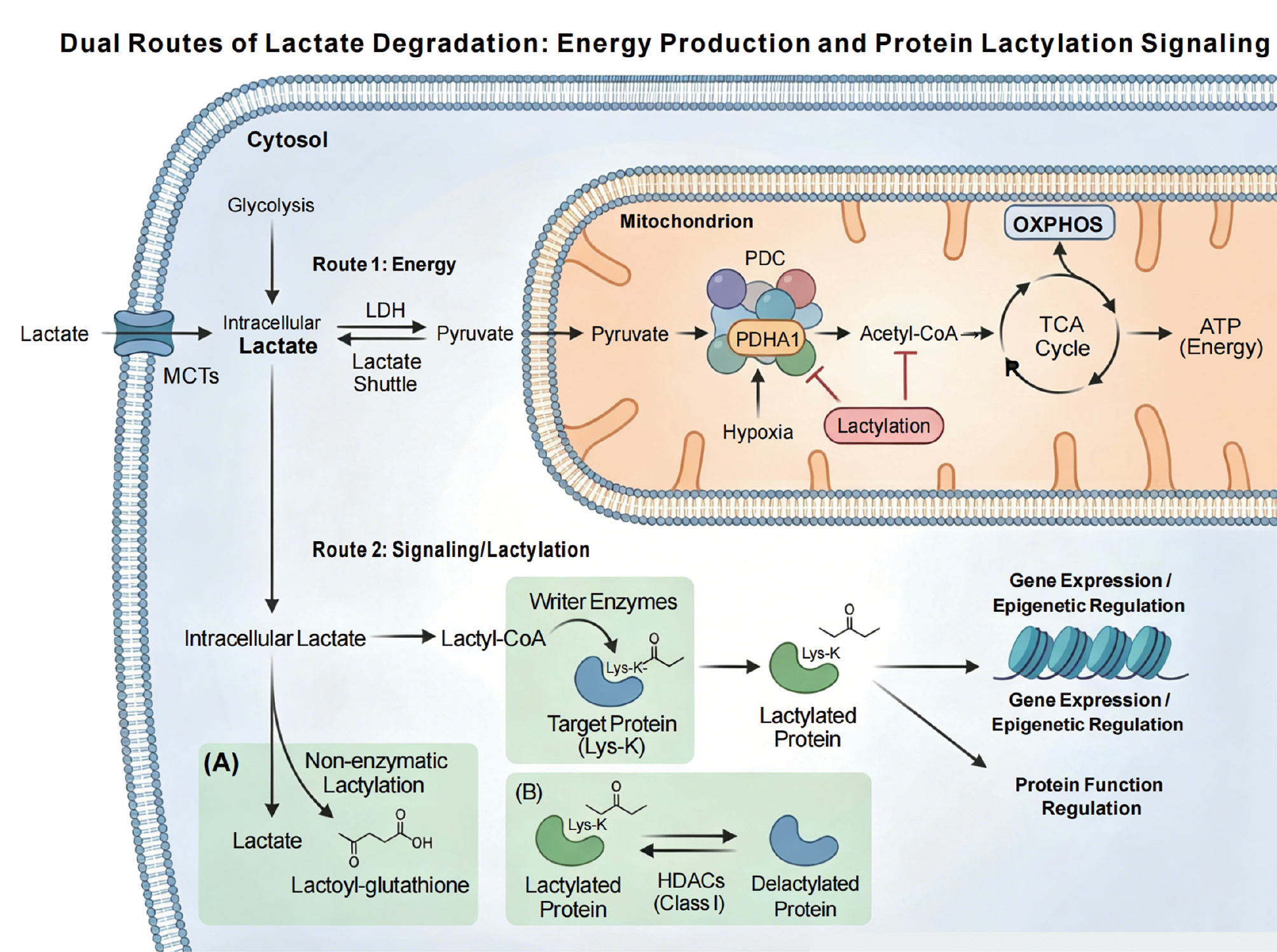
Lactate–pyruvate–TCA pathway
In the first route, lactate is reversibly oxidized to pyruvate by lactate dehydrogenase, the central step of the “lactate shuttle” between cytosol and mitochondria [35, 36]. The resulting pyruvate then enters mitochondria and is converted to acetyl-CoA by the pyruvate dehydrogenase complex (PDC), feeding into the TCA cycle and producing ATP efficiently via oxidative phosphorylation [26, 36]. Notably, this process is subject to fine regulation by lactylation. Recent studies have shown that under hypoxic conditions, lactylation of PDHA1 (the E1α subunit of PDC) directly inhibits the enzymatic activity, thereby limiting acetyl-CoA generation and subsequent oxidative phosphorylation, achieving a dynamic match between metabolic flux and oxygen supply [38].
Lactyl-CoA and protein lactylation
The second route reveals the role of lactate as a signaling molecule. Lactate can be converted into the high-energy intermediate, lactyl-CoA, which under the catalysis of specific “writer” enzymes, covalently transfers lactyl groups onto lysine residues of proteins to form lactylation [17, 26]. Lactyl-CoA has multiple sources. Lactyl-CoA may be synthesized in situ from intracellular lactate generated by glycolysis or arise from extracellular lactate imported via monocarboxylate transporters (MCTs), then converted intracellularly [17, 26]. In addition, lactylation mechanisms independent of lactyl-CoA have been described (e.g., non-enzymatic lysine lactylation via the intermediate lactoyl-glutathione and reversible lactylation/delactylation involving class I histone deacetylases [HDACs]) [26, 39]. These mechanisms collectively ensure that lactylation is highly sensitive to fluctuations in intracellular lactate, encoding metabolic states into epigenetic and protein-functional changes [17, 26, 40].
Lactate shuttle
The “lactate shuttle” theory systematically describes the dynamic transport and exchange of lactate between different cells, tissues, and even organs [41]. The core concept is that lactate serves as an important energy substrate and gluconeogenesis precursor and as a signaling molecule with extensive biological influence [35, 42].
The lactate shuttle is comprised of two principal types: intercellular shuttling; and intracellular shuttling. Intercellular shuttling denotes directed movement of lactate between cells or tissues, a process dependent on plasma-membrane monocarboxylate transporters, especially MCT1 and MCT4 [43]. Under physiologic conditions, such as exercise, “producer” skeletal muscle generates large amounts of lactate via glycolysis. Lactate is then transported through the circulation to “consumer” cells (heart, brain, and kidneys) and utilized as an important oxidative substrate, forming a body-wide organ-to-organ lactate shuttle network [44]. Microcircuits also exist within organs. For example, astrocytes take up glucose in the brain and metabolize glucose to lactate, which is then transported via MCTs to neurons to fuel neuronal activity, the well-known astrocyte–neuron lactate shuttle [45]. Analogous shuttles have been observed under pathologic conditions. Metabolically reprogrammed, glycolytic-activated fibroblasts export lactate via MCT1 to cardiomyocytes, which promotes cardiac hypertrophy, a fibroblast–cardiomyocyte lactate shuttle [46]. Shuttling is even more active in the tumor microenvironment. Highly glycolytic cancer cells or cancer-associated fibroblasts export lactate via MCT4, while neighboring oxidative cancer cells import lactate through MCT1, convert lactate to pyruvate, and channel lactate into the TCA cycle, a metabolic symbiosis that fosters tumor growth and immune evasion [43].
Intracellular lactate shuttling focuses on the movement and metabolism of lactate between subcellular compartments, particularly transfer from the cytosol to mitochondria [44]. Contrary to the traditional view that pyruvate is the main mitochondrial input derived from glycolysis, current studies have clearly shown that cytosolic lactate can also directly enter mitochondria [44], which is likely mediated by MCTs on the mitochondrial membrane. Once in the matrix, lactate is re-oxidized to pyruvate by mitochondrial LDH, then enters the TCA for energy production. This intracellular shuttle bears important physiologic significance. For example, anti-inflammatory M2 polarization coincides with increased mitochondrial localization of MCT1, facilitating lactate influx to support reparative functions. Blocking MCT1 disrupts mitochondrial function and drives a shift toward pro-inflammatory M1 polarization [47]. In CD8+ T cells, MCT1-mediated lactate efflux is critical for maintaining intracellular pH and glycolytic flux, which is essential for robust expansion and memory formation. MCT1 deficiency causes intracellular acidification and metabolic collapse, culminating in T-cell exhaustion [48].
These two shuttles are tightly interconnected. Intercellular shuttling supplies sources and sinks for intracellular shuttling. For example, lactate taken up by cancer cells from the microenvironment can be directly oxidized in mitochondria or reshape gene expression through epigenetic modifications, such as histone lactylation, thereby driving tumor progression. Conversely, the internal metabolic status of a cell determines whether the cell is a net lactate producer or consumer, thus defining the role within the intercellular shuttle network [44]. A deeper understanding of lactate shuttling offers new therapeutic targets. Inhibiting MCT1 or MCT4 can disrupt tumor metabolic symbiosis, reverse the immunosuppressive microenvironment, and potentially enhance immunotherapy [49, 50]. Such strategies may also mitigate synovial hyperplasia in rheumatoid arthritis [51] or suppress hypertensive cardiac remodeling [46]. Notably, MCT1 also has protective roles in the CNS energy supply [45], peripheral nerve regeneration [52], and anti-infective immunity [48]. Systemic inhibition of MCT1 could therefore have complex consequences. Precise, context- and cell type–specific modulation of lactate shuttling is thus a key direction for translational medicine.
System-level metabolic roles of lactate
The importance of lactate metabolism is reflected at least in three aspects at the system level: (1) a major recyclable energy source; (2) a key carbon precursor for gluconeogenesis; and (3) a signaling molecule with autocrine, paracrine, and endocrine-like effects, which is sometimes termed a “lactormone” [35, 53]. Figure 3 illustrates the system-level metabolic roles of lactate, highlighting how lactate shuttling, gluconeogenesis, and signaling pathways interconnect across various tissues and cellular contexts.
Figure 3 Systemic metabolic functions of lactic acid. Lactic acid has three core roles in the body. 1. Energy carrier: Lactic acid is transported between lactate-producing cells (e.g., tumor cells and skeletal muscles) and energy-consuming cells through the “lactate shuttle” mediated by monocarboxylate transporters (e.g., astrocyte-neuron lactate shuttle), where lactic acid is oxidized for energy production. 2. Glycogenesis precursor: Lactic acid produced in peripheral tissues is taken up by the liver via the bloodstream and converted into glucose through the Cori cycle, which is crucial for maintaining blood glucose homeostasis. 3. Signaling molecule: Lactic acid can serve as a “lactate hormone.” Lactic acid regulates lipolysis and inflammation by activating the GPR81 receptor. Lactic acid drives the lactylation modification of histones and non-histone proteins as a lactoyl donor, thereby directly coupling metabolic state with epigenetic regulation and gene expression.
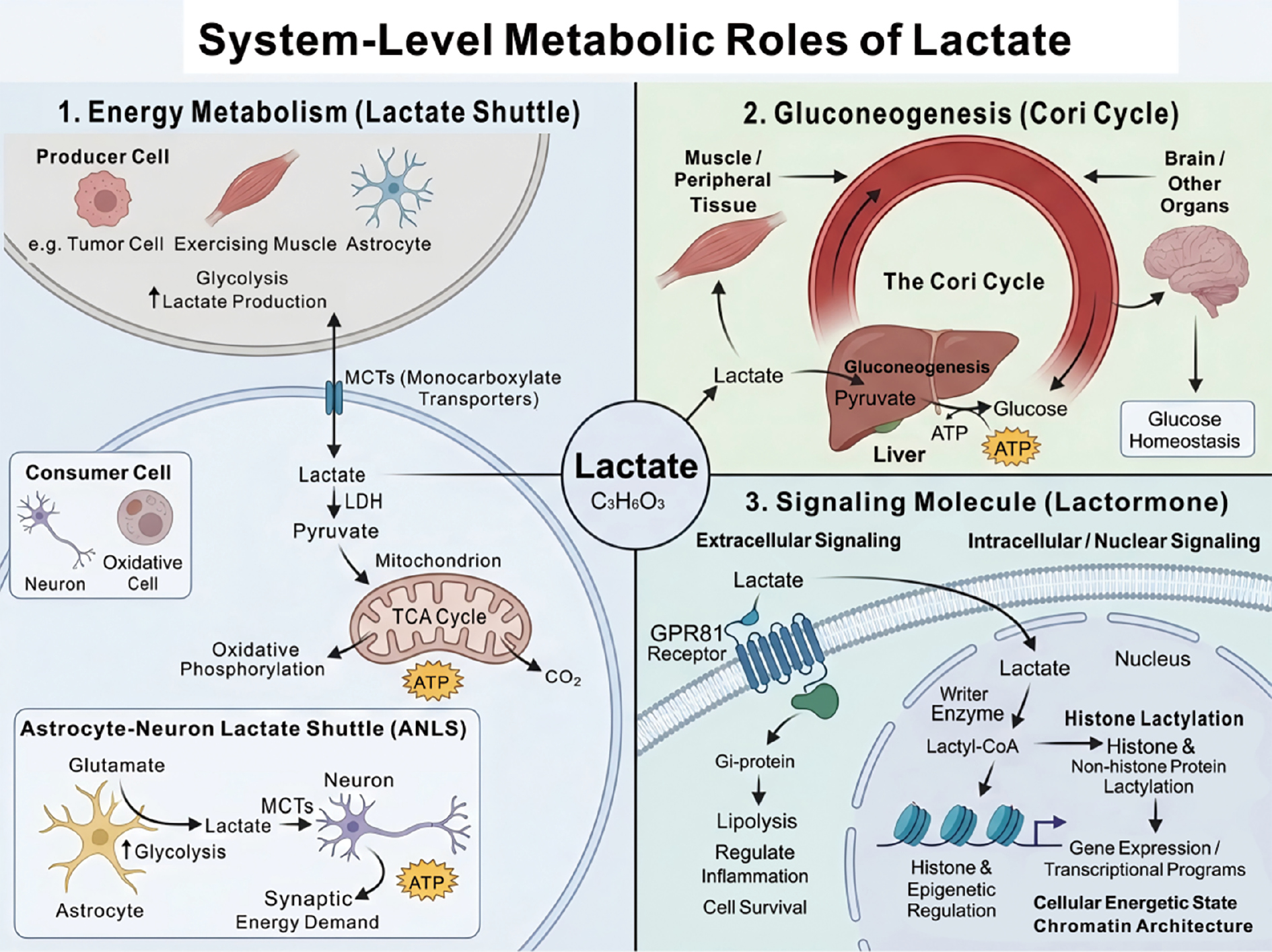
Energy metabolism
As an efficient circulating metabolite and energy carrier, lactate is produced and released in large amounts by glycolytically active cells (e.g., tumor cells, activated immune cells, or exercising skeletal muscle) and is subsequently taken up by neighboring or distant cells with different metabolic demands to serve as a substrate for oxidative phosphorylation [26, 35]. This “lactate shuttle,” which is mediated by MCTs, delivers lactate from “energy-donor” cells to “energy-consumer” cells, enabling metabolic cooperation and energy redistribution across cells and tissues [35, 36]. For example, the astrocyte–neuron lactate shuttle (ANLS) in the central nervous system is thought to couple synaptic activity with energy supply. Astrocytes enhance glycolysis and produce lactate in response to neurotransmitters, such as glutamate. Lactate is then transported via MCTs to neurons and oxidized to meet the high energy demands [35, 54]. Cancer-derived lactate not only contributes to local acidification and immune suppression in tumor microenvironments but is also utilized by cancer-associated fibroblasts or endothelial cells (“metabolic collaborators”) to drive reprogramming and angiogenesis, thereby indirectly supporting tumor growth and metastasis [25]. In high-demand tissues, such as the brain and retina, cells exhibit pronounced aerobic glycolysis and continuous lactate production even under oxygenated conditions [35, 54]. Rather than a mere waste, this lactate is shuttled and oxidized after conversion to pyruvate to fuel the TCA cycle and ATP generation in oxidative cells (e.g., neurons), which meets sustained and fluctuating energy needs [26, 35]
Gluconeogenesis
Lactate is a principal precursor for gluconeogenesis, particularly in the liver. Large amounts of lactate produced by peripheral tissues during exercise or stress, such as skeletal muscle, enter the circulation. The lactate is then taken up by hepatocytes and reconverted to glucose via gluconeogenesis, which is then released into the bloodstream to fuel the brain and other organs [36, 37] in the classic Cori cycle, an essential metabolic loop for maintaining glucose homeostasis. Isotope-tracing studies have further indicated that the carbon contribution of lactate to hepatic gluconeogenesis from fasting rest to moderate exercise can match or even exceed that of other traditional substrates [37]. Lactate may also be converted in immune cells, such as macrophages, into storage forms, like glycogen, which provides an energy reserve for effector functions in glucose-deprived inflammatory microenvironments [26].
Signaling molecule
In addition to the role of lactate as an energy substrate, lactate functions as a multimodal signaling molecule. Lactate activates Gi-protein–coupled downstream pathways to regulate lipolysis, inflammatory responses, and cell survival in various physiologic and pathologic situations by binding to a specific G-protein-coupled receptor GPR81 (also known as HCAR1) [53, 55]. In contrast, once entering the nucleus, lactate can be converted to lactyl-CoA and serve as an acyl donor for histone and non-histone protein lactylation. This process establishes a metabolism-dependent epigenetic regulatory mode that directly couples cellular energetic state to chromatin architecture and transcriptional programs [17, 26].
Lactate in PF
Fibroblasts
Extensive studies and pathologic observations indicate that the abundance of pulmonary fibroblasts and the formation of “fibroblastic foci” correlate closely with disease progression and poor outcomes, making fibroblasts the principal effector cell population in PF [2, 56–58]. These fibroblasts may originate from multiple lineages, including resident interstitial fibroblasts, pericytes, epithelial-to-mesenchymal transition, and circulating fibrocyte-like cells, exhibiting marked heterogeneity in origin and function [56, 57, 59]. Under stimuli, such as TGF-β, PDGF, and mechanical tension, fibroblasts differentiate into alpha-smooth muscle actin (α-SMA)–positive myofibroblasts that produce large amounts of collagen and fibronectin, leading to irreversible remodeling of the lung interstitium [56, 57, 60].
Recent studies have demonstrated that fibroblasts in fibrotic diseases are not merely passive structural effector cells. In fact, fibroblasts undergo a tumor-like metabolic reprogramming that is characterized by a shift from mitochondrial oxidative phosphorylation to aerobic glycolysis and is accompanied by increased lactate production [10, 60, 61]. Xie et al. reported that TGF-β-induced myofibroblast differentiation is critically dependent on enhanced glycolytic flux and upregulation of key glycolytic enzymes in murine models of PF and human lung fibroblasts, whereas pharmacologic inhibition of glycolysis using 2-deoxyglucose markedly attenuates experimental fibrosis [10]. Kottmann et al. also reported significantly elevated lactate levels in bronchoalveolar lavage fluid and lung tissue from patients with idiopathic PF with fibroblasts identified as a major source and lactate concentrations positively correlating with disease severity [11]. Together, these findings suggest that lactate is not merely a metabolic byproduct of enhanced glycolysis, but may function as an active pro-fibrotic signaling molecule that directly contributes to disease progression (Figure 4).
Figure 4 Fibroblasts in pulmonary fibrosis. Fibroblasts from multiple sources differentiate into α-SMA-expressing myofibroblasts under the influence of TGF-β, PDGF, and mechanical traction. Concurrently, mitochondrial oxidative phosphorylation declines, glycolysis is enhanced, and glycolytic enzymes, such as HK2, are upregulated. Lactate is excreted via MCT and accumulates extracellularly. The lactate-mediated microenvironmental acidification activates latent TGF-β, forming a feedback loop that continuously drives myofibroblast activation and matrix contraction, ultimately leading to excessive deposition of collagen and fibronectin, resulting in pulmonary fibrosis.
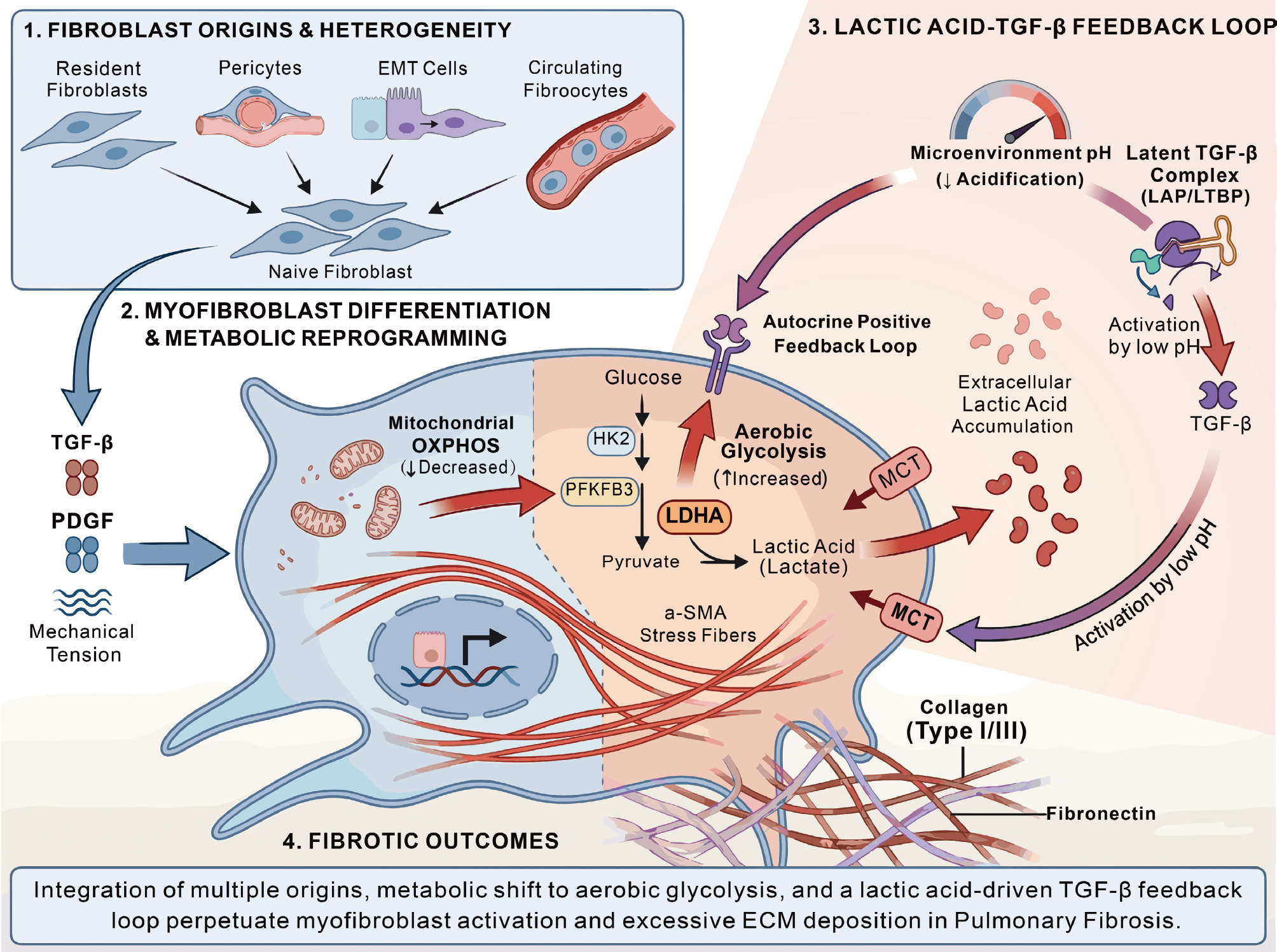
Mechanistically, the pro-fibrotic effects of lactate on fibroblasts can be conceptualized as two partially independent yet interconnected pathways. The first pathway is a pH-dependent mechanism, in which lactate-driven acidification of the microenvironment amplifies pro-fibrotic signaling. The second pathway is a lactylation-dependent epigenetic mechanism, in which lactate serves as a substrate for histone lactylation and related modifications, thereby reshaping transcriptional programs.
Enhanced glycolysis in fibroblasts leads to sustained lactate accumulation and a consequent reduction in local extracellular pH in fibrotic lung tissue [11]. Importantly, this acidification is not merely a metabolic byproduct but rather a functionally relevant signaling event. Lactate-induced acidification has been shown to promote the activation of latent TGF-β, thereby establishing a self-amplifying autocrine loop characterized by “lactate accumulation-microenvironment acidification-TGF-β activation,” which continuously drives myofibroblast differentiation and collagen synthesis [11]. In turn, TGF-β further upregulates key glycolytic enzymes, including lactate dehydrogenase A, thereby enhancing lactate production and reinforcing this positive feedback circuit [11, 60, 61]. In support of this model, Bernard et al. demonstrated that TGF-β stimulation enhances glycolytic activity and lactate production in fibroblasts, while simultaneously promoting stress fiber formation and matrix contraction, which indicated that lactate metabolism is closely linked not only to ECM synthesis but also to the contractile and mechanical functions of myofibroblasts [61]. In addition, the hypoxic microenvironment that is characteristic of fibrotic lung tissue stabilizes hypoxia-inducible factor-1alpha (HIF-1α), further promoting glycolysis and lactate production, and synergizing with TGF-β signaling to exacerbate fibroblast activation and matrix deposition [59, 62]. Thus, the defining feature of this pathway is that lactate indirectly enhances TGF-β activation and downstream pro-fibrotic signaling by altering the local acid–base environment.
In addition to a role in microenvironmental acidification, lactate can also act as a metabolic substrate that directly participates in epigenetic regulation. Emerging evidence indicates that lactate promotes transcriptional activation of inflammation- and fibrosis-related genes through mechanisms, such as histone lactylation [17]. In contrast to the pH-dependent pathway, this mechanism does not primarily rely on extracellular acidification but rather highlights lactate as a signaling metabolite that directly regulates nuclear transcriptional processes. From this perspective, lactate should no longer be viewed solely as the end-product of glycolysis but as a key mediator linking metabolic reprogramming to gene expression remodeling. Functionally, this link may enable fibroblasts to maintain a persistently activated phenotype, retaining high levels of collagen synthesis, inflammatory amplification, and pro-fibrotic transcriptional activity even after the initial stimuli have subsided [17]. Therefore, lactylation-dependent epigenetic regulation underscores the role of lactate in sustaining fibroblast activation and establishing a form of “transcriptional memory,” representing a critical extension beyond the traditional view that lactate acts primarily through acidification.
Taken together, these two mechanistic axes suggest that targeting fibroblast glycolysis, lactate production, and downstream signaling pathways may provide novel therapeutic opportunities for PF. Inhibition of glycolysis or lactate dehydrogenase activity reduces lactate levels, attenuates TGF-β signaling, suppresses myofibroblast differentiation, and decreases collagen deposition and fibrotic remodeling in multiple in vitro and in vivo models [11, 60, 61]. From a metabolic perspective, targeting lactate-associated pathways may complement existing anti-fibrotic therapies, such as nintedanib and pirfenidone, thereby offering additional therapeutic strategies for PF [2, 60, 61, 63, 64]. However, it is important to note that lactate also has essential physiologic roles in normal wound healing and immune regulation. Therefore, excessive or non-specific inhibition of lactate metabolism may impair tissue repair and disrupt immune homeostasis. Future therapeutic strategies will thus require precise modulation of lactate-related pathways with careful consideration of timing, dosage, and cell-type specificity [13, 17].
Epithelial cells
Within the multifactorial pathogenesis underlying PF, AECs have a central, nodal role [65, 66]. AECs are not only the initial targets of injury but also key drivers of disease progression [67]. Under normal conditions, AECs, especially the regenerative AEC2 population, maintain alveolar structural integrity and microenvironmental homeostasis by secreting surfactant and serving as epithelial progenitors. Repetitive microinjury leads to severe epithelial dysfunction in PF. Repair and regeneration are impaired, while a series of pro-fibrotic programs are initiated and sustained, transforming epithelial cells from “repairers” into “engines” of fibrosis [68]. A core dysfunctional phenotype is metabolic reprogramming. AEC2s shift from mitochondrial oxidative phosphorylation to a high reliance on aerobic glycolysis, accompanied by markedly increased lactate production and local accumulation [12, 67].
Epithelial cell–derived lactate should not be regarded as a simple metabolic “waste product” but rather as a critical signaling molecule and pathologic mediator that contributes to PF progression through multiple mechanisms.
Enhanced glycolysis in epithelial cells leads to sustained lactate accumulation in fibrotic lung tissue with local microenvironmental acidification as one of the most immediate consequences [12]. Importantly, this acidification is not a passive bystander effect but a biologically active process that significantly influences epithelial cell fate and intercellular signaling. Accumulating evidence suggests that high lactate concentrations or an acidic microenvironment can directly induce EMT in AECs, resulting in loss of epithelial polarity, acquisition of mesenchymal features, and upregulation of multiple fibrosis-associated markers, thereby promoting PF progression [12, 69]. In this context, the key role of lactate lies in an ability to destabilize epithelial identity and enhance pro-fibrotic transcriptional and secretory responses through modulation of the extracellular pH.
In addition to driving EMT, lactate accumulation is closely associated with epithelial injury and loss. Mechanistically, lactate has been shown to activate the endoplasmic reticulum (ER) stress–associated ATF4–CHOP signaling axis, leading to activation of caspase-12 and induction of apoptosis in AECs [70]. Pharmacologic inhibition of ER stress can effectively block this process and attenuate fibrosis in murine models [69]. From a pathologic perspective, this cascade contributes to alveolar structural disruption and loss of epithelial barrier integrity, representing a key early event in fibrogenesis. Although it remains to be fully clarified whether these effects are primarily driven by lactate or microenvironmental acidification, the effects are mechanistically aligned with a pH-dependent pathway, in which lactate accumulation amplifies local cellular stress and tissue injury.
In contrast to these acidification-dependent effects, lactate can also function as an epigenetic substrate that directly regulates transcriptional programs within epithelial cells. Recent studies have demonstrated that upon transport into cells via monocarboxylate transporter 1, lactate can be converted into lactyl-CoA and subsequently drive site-specific histone lactylation, thereby activating gene expression programs associated with EMT, inflammation, and fibrosis [71]. This mechanism emphasizes lactate is a signaling metabolite that modulates chromatin structure and transcriptional activity independent of extracellular pH changes.
Lactate accumulation has been shown to promote histone H3K18 lactylation in in silica-induced models of PF, leading to upregulation of the transcription factor, SIX1, and subsequent induction of EMT in AECs, thereby accelerating fibrotic progression [72]. Similarly, lactate secreted by myofibroblasts can be taken up by AECs via MCT1 in arsenic-induced fibrosis models, inducing intracellular H3K18 lactylation and upregulating the m6A reader protein YTHDF1. YTHDF1, in turn, enhances the translation of the pro-fibrotic factor, NREP, and promotes TGF-β1 secretion, which further drives fibroblast-to-myofibroblast differentiation and ultimately establishes a positive feedback loop between epithelial cells and fibroblasts [71]. These findings indicated that lactylation-dependent mechanisms not only regulate epithelial cell-intrinsic phenotypic remodeling but also amplifies local pro-fibrotic signaling networks through post-transcriptional and paracrine pathways.
Moreover, lactylation-driven epigenetic reprogramming may also mediate aberrant crosstalk between epithelial cells and the immune microenvironment. Stromal cells exposed to stimuli in fibrotic and injury settings, such as TGF-β1, exhibit enhanced glycolysis and lactate release. This lactate can induce histone lactylation in macrophages, reprogramming the phenotype and promoting expression of multiple pro-fibrotic mediators, thereby further amplifying the local inflammation–fibrosis signaling network [12, 73]. Thus, a defining feature of the lactylation-dependent pathway is the ability to establish a sustained pro-fibrotic “transcriptional memory” or “state maintenance” program across epithelial and microenvironmental cell populations.
In addition to these two principal mechanisms, lactate accumulation is also closely associated with broader metabolic dysregulation in epithelial cells, forming an important background for the persistent progression of PF. Elevated lactate levels are frequently accompanied by lipid metabolic abnormalities, including increased lipid droplet accumulation within epithelial cells [74]. The molecular chaperone, CCT6A, has been shown to promote ubiquitin-mediated degradation of HIF-1α, thereby suppressing HIF-1α–driven glycolysis and lactate production, and ultimately reducing lipid accumulation and fibrosis severity [74]. Conversely, downregulation of ACSS3 impairs fatty acid oxidation while enhancing glycolysis, leading to increased lactate levels and subsequent mitochondrial dysfunction, elevated reactive oxygen species (ROS) production, impaired mitophagy, and apoptosis, collectively exacerbating epithelial cell dysfunction and fibrotic progression [75]. These findings suggested that lactate metabolic dysregulation is not an isolated event but rather part of a broader pathologic reprogramming network involving mitochondrial injury, lipid metabolic imbalance, and disrupted cell fate regulation in epithelial cells.
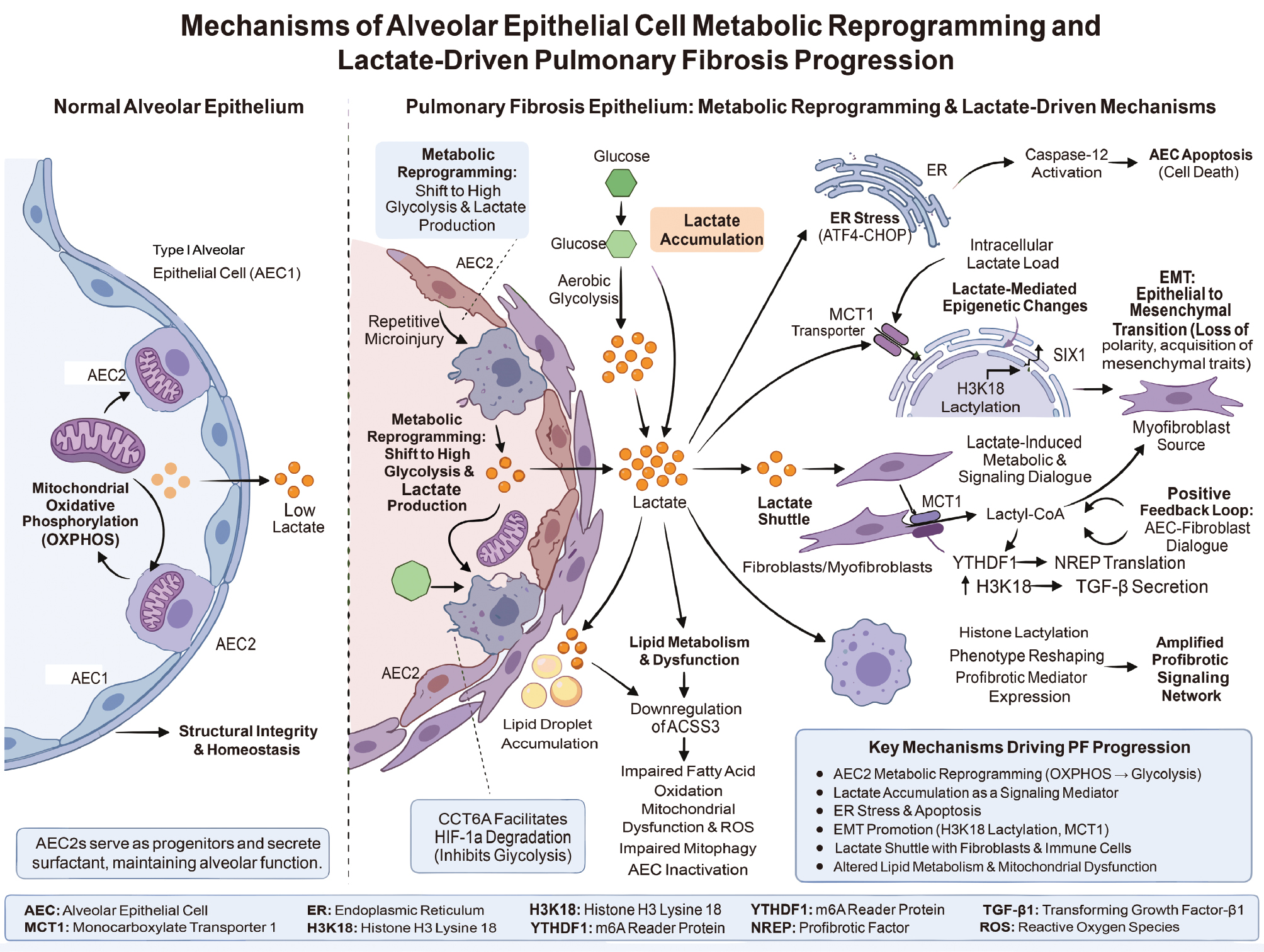
In sum, metabolic reprogramming of AECs and consequent lactate accumulation constitute key mechanistic links driving PF onset and progression. Strategies ranging from inhibiting aberrant glycolysis and blocking lactate production/transport to targeting lactate-related epigenetic modifications (e.g., histone lactylation) hold promise for PF prevention and therapy [12, 71]. Figure 5 provides a schematic illustration of the mechanisms underlying AEC metabolic reprogramming, lactate accumulation, and the contribution to PF progression.
Figure 5 Schematic diagram of metabolic reprogramming in alveolar epithelial cells and the mechanism of lactate-driven pulmonary fibrosis. In normal alveoli, type II alveolar epithelial cells rely on mitochondrial oxidative phosphorylation for energy supply and secrete surfactants to maintain structural integrity and homeostasis with minimal lactate production. Repeated microinjuries induce AEC2 to shift from OXPHOS to high glycolysis during pulmonary fibrosis, leading to substantial lactate generation, accompanied by lipid metabolism disorders, lipid droplet accumulation, and mitochondrial dysfunction. Accumulated lactate exerts dual effects. Lactate triggers endoplasmic reticulum stress, apoptosis, and epithelial-to-mesenchymal transition through transporters, such as MCT1 and histone acylation, forming new fibroblasts (myofibroblasts). In contrast, lactate mediates the “lactate shuttle” between fibroblasts, amplifying fibrotic signals, such as TGF-β and cytokine expression, thereby promoting collagenous extracellular matrix deposition and advancing pulmonary fibrosis.
Macrophages
Macrophages also serve as central drivers in the complex PF pathology [76]. In addition to acting as “sentinels” of inflammation, macrophages connect tissue injury, metabolic reprogramming, and fibrotic progression [77, 78]. Shifts in the functional state of macrophages, particularly a tilt toward glycolysis and the resultant lactate accumulation, have been validated as important mechanisms promoting PF development [24, 79, 80]. Macrophage roles in PF are multilayered. Alveolar macrophages in the fibrotic lung often exhibit a unique pro-fibrotic M2-like phenotype that does not fully overlap with classical M1/M2 categories [80]. Activated macrophages directly stimulate fibroblast-to-myofibroblast transition and excessive ECM deposition by secreting large amounts of TGF-β1, PDGFA, etc [81, 82].
Importantly, metabolic reprogramming in macrophages and the major metabolic product (lactate) constitute a central metabolic axis driving fibrosis. Macrophages consistently exhibit enhanced glycolysis in PF across multiple contexts, including idiopathic pulmonary fibrosis (IPF) patients and animal models induced by bleomycin, silica nanoparticles, or PM2.5 exposure [24, 80, 83]. This metabolic shift is not merely an adaptive response but is required for the acquisition of a pro-fibrotic phenotype. Indeed, alveolar macrophages in fibrotic lungs rely on glycolysis rather than fatty acid oxidation or glutaminolysis to sustain an M2-like pro-fibrotic state. Inhibition of glycolysis correspondingly attenuates the pro-fibrotic activity [80, 84].
Mechanistically, the pro-fibrotic effects of lactate on macrophages can be conceptualized along two partially independent yet interconnected axes. The first pathway is a pH-dependent pathway, in which lactate-induced acidification modulates macrophage activation and intercellular communication. The second pathway is a lactylation-dependent epigenetic pathway, in which lactate directly reshapes pro-fibrotic gene expression programs via histone lactylation and related modifications. Notably, current evidence more strongly supports the latter mechanism in macrophages, whereas the former appears to act primarily through indirect modulation of the fibrotic microenvironment.
Macrophages function both as a major source of lactate and as key responders to lactate signaling within fibrotic lesions. Activated macrophages produce substantial amounts of lactate due to enhanced glycolysis, while activated fibroblasts and myofibroblasts further contribute to lactate accumulation through increased glycolytic flux, collectively elevating local lactate concentrations [82]. This accumulation promotes microenvironmental acidification, thereby influencing signaling interactions among macrophages, fibroblasts, epithelial cells, and other immune populations. From a pathologic perspective, such acidification favors the persistence of chronic inflammation and dysregulated repair within fibrotic niches. For example, macrophage-derived TNF-α can activate PFKFB3 in lung fibroblasts, enhancing aerobic glycolysis and lactate production, thus amplifying local lactate accumulation and fibrotic progression [81]. Sustained secretion of chemokines, such as CCL2 and CXCL10, by macrophages promotes continuous recruitment of monocytes/macrophages and other inflammatory cells, including neutrophils, lymphocytes (e.g., T cells), and dendritic cells, to the injured site [78, 85]. In this context, lactate and the associated acidification act primarily as an amplifier, enhancing immune cell recruitment, reinforcing metabolic coupling between cell types, and maintaining a pathologic repair niche. However, the evidence supporting a direct role of low pH in dictating macrophage pro-fibrotic phenotypes remains relatively limited compared to fibroblasts, in which acidification directly activates latent TGF-β.
In contrast, the lactylation-dependent pathway represents a more direct and well-defined mechanism through which lactate drives macrophage pathogenicity. Lactate can enter the nucleus and with the involvement of enzymes, such as p300, promote histone lactylation at specific loci, particularly H3K18, thereby directly activating transcription of multiple pro-fibrotic genes [82, 83]. This mechanism highlights lactate as not merely a metabolic indicator but as an epigenetic substrate that converts metabolic alterations into sustained activation of fibrotic gene expression programs. Lactate-induced histone lactylation has been shown to enrich at the promoters of genes, such as ARG1, PDGFA, THBS1, and VEGFA, which enhances the transcriptional output [82]. H3K18 lactylation directly regulates key genes in silica nanoparticle–induced models of PF, such as NOS2 in macrophages, promoting inflammatory responses and fibrotic progression [83]. Similarly, enhanced glycolysis and subsequent increases in histone lactylation in macrophages have been identified as key metabolic–epigenetic drivers of the EMT and fibrosis in PM2.5 exposure models [24]. These findings indicate that lactylation is not a peripheral modification in macrophages, but rather a central molecular mechanism underpinning their pro-fibrotic activation.
In addition to transcriptional regulation, lactylation may also influence protein stability and degradation pathways, thereby further amplifying pro-fibrotic signaling. For example, elevated lactate levels in macrophages promote H3K18 lactylation at the promoter region of Stub1, leading to suppression of the E3 ubiquitin ligase, CHIP, in PM2.5-induced PF models. Given that CHIP mediates ubiquitination and degradation of TGF-β1, downregulation of CHIP results in increased stability and secretion of TGF-β1, ultimately exacerbating fibrosis [79]. This finding suggests that lactylation-dependent mechanisms can regulate gene transcription and proteostasis networks, thereby sustaining the presence of key pro-fibrotic mediators.
Macrophage polarization is tightly coupled to lactate metabolism and forms a self-reinforcing loop in PF. A lactate-enriched microenvironment promotes macrophage polarization toward an M2-like pro-fibrotic phenotype, which in turn sustains glycolytic activation and continuous lactate production through the secretion of growth factors and inflammatory mediators [86]. For example, the m6A reader protein, IGF2BP1, stabilizes THBS1 mRNA, activates Toll-like receptor 4-associated signaling, and promotes macrophage glycolysis and M2 polarization, thereby accelerating fibrotic progression [86]. Conversely, inhibition of glycolysis in macrophages reduces glycolytic flux, attenuates the M2-like program, and ameliorates PF [84]. These observations indicate that macrophages are not passive responders to lactate signals but actively establish a closed-loop system linking metabolic reprogramming, phenotypic polarization, and paracrine signaling. In this sense, lactate is both a product of macrophage activation and a driver of a sustained pro-fibrotic phenotype. Accordingly, the macrophage glycolysis–lactate axis is increasingly recognized as a core pathologic pathway in PF with both driving and maintenance functions [79, 82].
Based on these mechanisms, targeting the macrophage glycolysis–lactate axis has emerged as a promising therapeutic strategy. For example, the GLP-1 receptor agonist, liraglutide, has been shown to exert anti-fibrotic effects by disrupting the interaction between NLRP3 inflammasome activation and PFKFB3-driven glycolysis, thereby inhibiting lactate-mediated histone lactylation [87]. Collectively, strategies aimed at suppressing aberrant glycolysis in macrophages, reducing lactate production, or blocking lactate-driven histone lactylation and downstream transcriptional programs may represent novel and effective approaches for the treatment of PF.
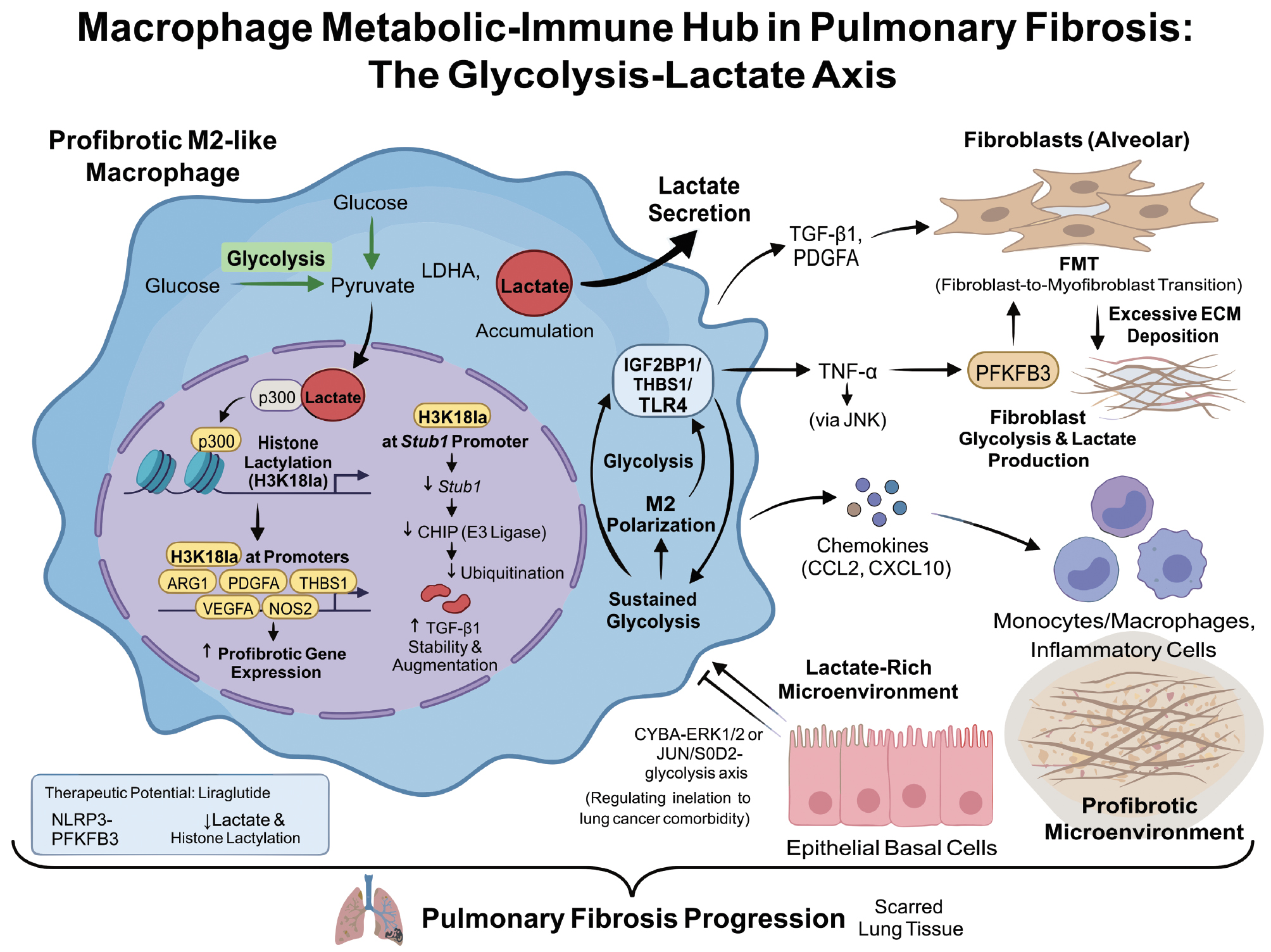
In conclusion, macrophages occupy a dual metabolic–immune hub in PF with functional abnormalities tightly linked to metabolic reprogramming. The increase in lactate arising from enhanced glycolysis is not a simple byproduct but by inducing histone lactylation, stabilizing profibrotic factors, and modulating macrophage polarization, a powerful pro-fibrotic signaling network is constructed [79, 82]. Figure 6 illustrates the glycolysis-lactate axis in macrophages, highlighting the role of lactate in modulating macrophage function, promoting fibrosis, and sustaining a pro-fibrotic microenvironment in PF.
Figure 6 Schematic diagram of macrophage function in pulmonary fibrosis. Fibrosis-promoting M2-like macrophages exhibit enhanced glycolysis, in which pyruvate is extensively converted to lactic acid under LDHA catalysis and accumulates. Lactic acid enters the nucleus and undergoes p300-mediated histone H3K18 acetylation, activating fibrosis-promoting genes, such as ARG1, PDGFA, THBS1, VEGFA, and NOS2, while suppressing Stub1/CHIP expression, thereby stabilizing and amplifying the TGF-β1 signaling pathway. Macrophages simultaneously secrete factors, including lactic acid, TGF-β1, PDGFA, and TNF-α, driving fibroblast glycolysis and lactic acid production, fibroblast-to-myofibroblast transdifferentiation, and excessive ECM deposition. Additionally, they recruit more inflammatory cells through chemokines such as CCL2 and CXCL10, forming a lactic acid-enriched and fibrosis-promoting microenvironment that advances pulmonary fibrosis. The GLP-1 receptor agonist liraglutide demonstrates potential anti-fibrotic effects by inhibiting the NLRP3-PFKFB3 pathway and reducing lactic acid and histone acetylation.
Lactylation
Lactylation is a recently discovered post-translational modification that directly links the metabolic product, lactate, to protein functional regulation, marking a new stage in our understanding of the interplay between cellular metabolism and gene expression [17]. In essence, this modification covalently attaches a lactyl group derived from lactate to lysine residues of proteins, thereby altering protein structure, function, stability, and interaction networks [41, 88]. This section systematically elaborates the molecular mechanisms and key regulatory factors of lactylation, including the enzymes and proteins that catalyze, remove, and recognize this modification, as well as diverse functions in physiologic and pathologic processes.
Lactylation regulatory mechanisms
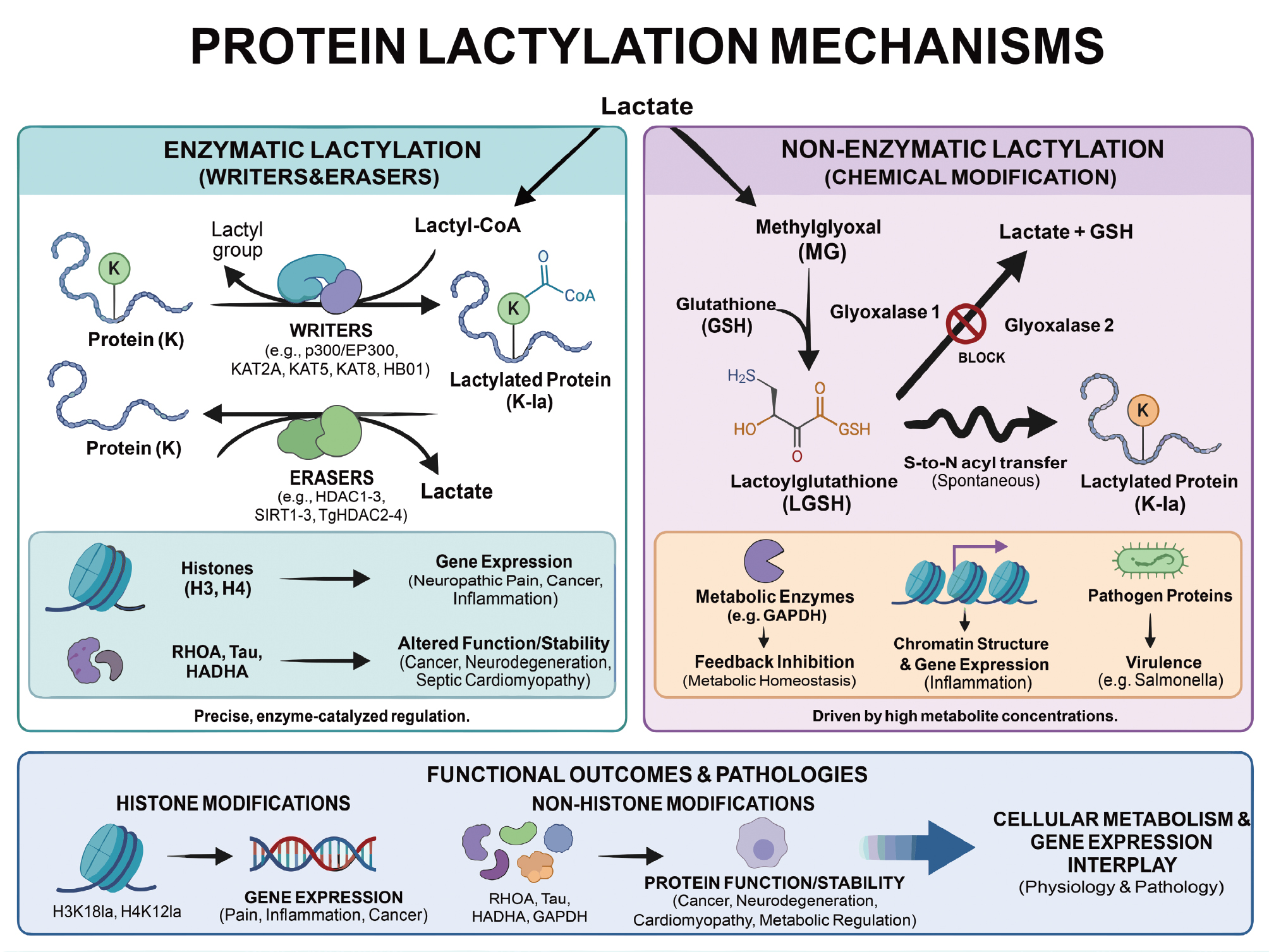
As a key post-translational modification, lactylation has an increasingly important role in regulating cellular processes. This modification can occur enzymatically or non-enzymatically and impacts various proteins involved in metabolism, gene expression, and cellular signaling [88]. A deeper understanding of these mechanisms can shed light on the dual roles of lactate in both normal physiologic processes and pathologic conditions, such as cancer and chronic inflammation. Figure 7 provides a comprehensive overview of the different lactylation mechanisms, distinguishing between the enzyme-catalyzed processes and those processes driven by metabolic intermediates. The figure illustrates how lactylation modifies proteins, regulates cellular functions, and contributes to the development of diseases, like fibrosis.
Figure 7 Schematic diagram of enzymatic and non-enzymatic mechanisms of lactic acidification and functional outcomes. The left panel illustrates enzymatic lactylation: Lactyl-CoA generated from lactic acid is modified by protein lysines under the catalysis of “writing enzymes,” such as p300/EP300, KAT2A, and KAT5, while “erasing enzymes,” like HDAC1-3 and SIRT1-3 remove the lactyl group, achieving dynamic and reversible regulation. Histone H3 and H4 lactylation regulates gene transcription, whereas non-histone lactylation alters protein conformation, stability, and enzymatic activity, participating in pathologic processes, such as inflammation, metabolic disorders, and tumors. The right panel depicts non-enzymatic lactylation: Glycolytic byproduct methylmalonate reacts with glutathione to form lactylglutathione under GLO1 catalysis. Abnormal accumulation occurs when GLO2 function is impaired, in which LGSH spontaneously modifies metabolic enzymes, histones, and pathogen proteins through S-N acetyl transfer, thereby feedback-regulating glycolysis and remodeling chromatin. The lower panel summarizes lactylation-mediated alterations in protein function and disease pathogenesis, as well as a pivotal role in the interplay between cellular metabolism and gene expression.
Enzymatic lactylation
Enzymatic lactylation is a newly recognized, enzyme-catalyzed PTM that covalently links a lactyl group to lysine residues of proteins, dynamically regulating protein function and activity [89]. This process is orchestrated by a refined enzymatic system.
Lactylation is dynamically controlled by “writers” and “erasers.” Multiple classical acetyltransferases have been shown to possess lactyltransferase activity, forming a diversified writer family [90]. For example, the canonical histone acetyltransferase, p300/EP300, catalyzes H3K18la and H4K12la in sensory neurons, thereby regulating the expression of neuropathic pain–related genes [91]. p300 is also a key enzyme for novel histone lactylation sites (H4K79la and H4K91la) in breast cancer [92]. Other acetyltransferases, such as KAT2A, KAT5, KAT8, and HBO1, exhibit similar lactyltransferase functions [93–96]. Removal of lactyl groups is carried out by specific deacetylases. Histone deacetylases (TgHDAC2–4) were first identified to possess delactylase activity and remove protein lactyl groups in Toxoplasma gondii [97]. Class I HDACs (HDAC1–3) and NAD+-dependent deacetylases (SIRT1–3) have also been implicated in mammals [40, 98]. The dynamic balance between writers and erasers precisely maintains intracellular lactylation levels and functions [99, 100].
Enzymatic lactylation influences protein conformation, catalytic activity, stability, and intermolecular interactions, thereby impacting broad physiologic and pathologic processes [101, 102]. Lactylation of RHOA at K118 and K162 impairs GTPase activity and competitively antagonizes ubiquitination in tumorigenesis, constitutively activating and stabilizing the protein to drive cancer progression [101]. p300-catalyzed lactylation of Tau at K331 in neurodegeneration enhances pathologic phosphorylation and cleavage, an association that occurs with Alzheimer’s disease pathology [103]. Lactylation of HADHA at K166 and K728 in septic cardiomyopathy suppresses HADHA enzymatic activity, which disturbs mitochondrial energy metabolism [102]. This modification also directly participates in transcriptional control. For example, glycolytic reprogramming driven by PFKFB3 increases lactate and enhances H4K12la, activating NF-κB–pathway genes and promoting renal inflammation and fibrosis [104]. Hence, insights into enzymatic lactylation not only illuminate metabolism–immunity crosstalk in chronic diseases but also open new therapeutic avenues.
Non-enzymatic lactylation
Non-enzymatic lactylation refers to spontaneous chemical modification of lysine residues in proteins driven by high concentrations of reactive metabolic intermediates that occur without specific writer enzymes [105]. The core driving force for non-enzymatic lactylation is abnormal accumulation of metabolites rather than enzymatic catalysis [39, 105].
The best characterized mechanism involves lactoylglutathione (LGSH). LGSH is a key intermediate in the glyoxalase detoxification pathway. The glycolytic byproduct, methylglyoxal, conjugates with glutathione and under glyoxalase 1 catalysis, forms LGSH [39, 106]. Normally, LGSH is rapidly hydrolyzed by glyoxalase 2 to D-lactate, while regenerating glutathione to complete the detoxification cycle [39]. LGSH accumulates abnormally when glyoxalase 2 is absent or inhibited [39, 106]. High concentrations of LGSH are highly reactive and can spontaneously transfer the lactyl group to neighboring lysine residues via S-to-N acyl transfer, generating lactylation [39, 106, 107]. Reaction efficiency is influenced by LGSH levels, local pH, and a redox environment [107]. LGSH can also transfer lactyl groups to coenzyme A via S-to-S acyl transfer to form lactyl-CoA, highlighting the diversity of lactyl donors [106].
Non-enzymatic lactylation targets are widespread and enriched among metabolic proteins. A prototypical regulatory outcome is reversible inhibition of key glycolytic enzymes, forming a novel feedback loop. For example, GAPDH can be modified in plants by LGSH, resulting in lactylation at multiple lysine sites and cysteinylation at key cysteine residues, thereby reversibly inhibiting enzymatic activity [107]. The physiologic logic is that excessive glycolysis generates more methylglyoxal and LGSH. Accumulated LGSH then suppresses rate-limiting glycolytic enzymes via non-enzymatic lactylation, which negatively regulates glycolytic flux to maintain metabolic homeostasis [39].
Non-enzymatic lactylation also occurs on histones and affects gene expression. LGSH accumulation due to glyoxalase 2 deficiency correlates with elevated histone lactylation and heightened inflammatory responses in macrophages. Interestingly, histone lactylation and acetylation may differentially impact chromatin architecture. At rest lactylation associates with more compact chromatin, whereas under inflammatory stimulation, chromatin accessibility at these regions increases, thereby regulating inflammatory gene expression [106]. In addition to metabolism and epigenetics, lactylation has important roles during pathogen infection. Specifically, lactylation (enzymatic and non-enzymatic) participates in epithelial invasion and is crucial for virulence in Salmonella [105].
The relative contributions, kinetics, and functional specificity of non-enzymatic versus enzymatic lactylation under physiologic and pathologic conditions remain frontiers of debate. One view posits that highly conserved, function-critical sites on core proteins, such as histones, may still require precise enzymatic control, whereas non-enzymatic modifications might occur more broadly on other proteins as a widespread signal amplifier. Dissecting the interplay of these mechanisms is essential for a comprehensive understanding of the dual roles of lactate in cellular signaling.
Lactate-oxidized write-erase-read system
Lactylation directly couples cellular metabolism to epigenetic regulation and protein function. The dynamic control of lactylation adheres to the classical “write–erase–read” paradigm [108], forming a precise signaling system that enables cells to sense microenvironmental changes, such as hypoxia and high glycolysis, and to have key roles in transcription, cell-fate decisions, and disease progression [109].
Writers: establishment of lactylation
Writers covalently attach lactyl groups to specific lysines on target proteins, which initiates the lactylation signaling cascade. Lactylation can arise via enzymatic and non-enzymatic mechanisms with the enzymatic route being the current focus [108]. In this route lactate is first activated into high-energy lactyl-CoA, potentially by a lactyl-CoA synthetase (the molecular identity of which in mammals has not been fully defined), and specific acyltransferases subsequently transfer lactyl groups to lysines.
Writers of histone lactylation are relatively well-studied. p300 was the first identified histone lactylation writer within the CBP/p300 HAT family [17, 108–110]. Overexpression or knockdown of p300 increases or decreases histone lactylation, respectively [17]. p300 is the principal writer catalyzing H3K18la in pancreatic ductal adenocarcinoma and is essential for malignant phenotypes [109]. p300 forms complexes with the chromatin remodeler, BRG1, and the eraser, HDAC3, to co-regulate H3K18la and metastasis-related gene expression in colorectal cancer liver metastasis models [110]. Interestingly, HDAC1/2/3 (classically considered deacylases) can reverse-catalyze lactylation under specific conditions and act as intracellular drivers of lactylation [40]. For example, HDAC3 has been reported to be a delactylase for NBS1, suggesting bidirectional regulation at that site [111].
Writers highlight the breadth and substrate specificity of lactylation for non-histone proteins. AARS1 was identified as the specific writer for K23 lactylation of the RNA-processing factor, NUDT21, in esophageal squamous cell carcinoma [112]. Acetyltransferase KAT8 functions as a “pan-lactylation writer” in colorectal cancer, catalyzing lactylation of multiple substrates, including eEF1A2. Lactylation at K408 promotes translational elongation and protein synthesis to meet rapid tumor growth demands [95].
In contrast, non-enzymatic routes involve direct chemical reactions between active lactyl donors, such as lactoylglutathione and protein lysines [108]. Such chemistry may operate under bacterial infection or metabolic stress. However, the regulatory specificity and biological significance are more complex compared to precise enzyme-mediated pathways and warrant further exploration.
Erasers: clearing the modification and maintaining balance
Erasers remove lactyl groups from lysines, which terminates lactylation signals, restores proteins, or frees sites for other PTMs. This process is essential for the dynamic balance and reversibility of lactylation. Erasers are mainly derived from the following two families: zinc-dependent HDACs; and NAD+-dependent sirtuins [21, 108].
Class I HDACs (HDAC1/2/3) are efficient delactylases in mammalian cells with critical roles across contexts [113, 114]. For example, HDAC2 serves as a specific eraser of H3K9la during angiogenesis and H3K9la suppresses HDAC2 expression in feedback, forming a sensitive loop controlling neovascularization [115]. HDAC3 removes H4K12la in macrophages to modulate downstream TGF-β signaling and collagen synthesis in dermal fibroblasts [116]. HDAC2 has likewise been identified as a potential histone delactylase in pancreatic ductal adenocarcinoma [109]. Moreover, hypoxia downregulates HDAC8, a specific delactylase for PRMT1, in breast cancer metastasis. The reduced HDAC8 elevates PRMT1 lactylation, enhances methyltransferase activity, and drives metastasis [117].
Sirtuins, particularly SIRT1–3 and SIRT6, also display delactylase activity with varying catalytic efficiencies and substrate selectivity [21, 114]. SIRT1 was the first reported sirtuin to delactylate histone H3K18la [118]. Genetically encoded probes further suggest potential activity on non-histone substrates [119]. SIRT1 delactylates the α-myosin heavy chain at K1897 in the heart to dynamically regulate the interaction with titin and thus cardiac function [120]. SIRT3 exhibits high selectivity for some histone sites. SIRT3 is a principal binder and specific eraser of H3K9la, which suppresses transcription and cancer progression in esophageal squamous cell carcinoma [121]. SIRT3 also exhibits strong activity toward H4K16la [114]. SIRT6, a key nuclear deacylase, demonstrates remarkable plasticity and can remove multiple metabolically related histone marks, including lactylation and β-hydroxybutyrylation [122]. Together, these erasers form a multilayered, finely tuned clearing system that ensures precision and dynamics of lactylation in signaling.
Readers: decoding the signal and executing effects
Readers specifically recognize and bind lactylation, converting a chemical mark into downstream biological effects. By sensing conformational or electrostatic changes introduced by lactyl groups, readers remodel chromatin, recruit transcriptional complexes, or regulate protein-interaction networks. Although nascent, several histone lactylation readers have been identified.
In 2024 the catalytic subunit (Brg1) of the SWI/SNF chromatin-remodeling complex was reported as a reader of H3K18la. Dux-induced H3K18la recruits p300 during induced pluripotent stem cell reprogramming and modulates a metabolism–H3K18la–MET network to initiate a “metabolic switch” and enhance reprogramming efficiency. Brg1 is specifically recruited by H3K18la, co-localizing at promoters of pluripotency and epithelial-junction genes to promote the mesenchymal-to-epithelial transition [123]. Another study identified DPF2 as a specific binder of H3K14la in cervical cancer cells, in which lactate-accumulation–driven H3K14la and DPF2 co-enrich at oncogenic promoters to drive transcription and tumor growth. Disrupting this interaction suppresses oncogene expression and viability [124]. In addition, the bromodomain protein, TRIM33, binds lactylated histone peptides with sub-micromolar affinity. A unique glutamate residue in the bromodomain confers specificity for lactylation. TRIM33 reads lactylation in macrophages to attenuate late inflammatory genes and modulate polarization [125].
“Reading” can be more diverse for non-histone proteins and may not always rely on separate reader proteins. The m6A reader, YTHDC1, undergoes self-lactylation at K82 in renal cell carcinoma, which enhances phase separation capacity rather than requiring an external reader. Lactylation promotes YTHDC1 condensate formation in the nucleus, which protects oncogenic transcripts (BCL2 and E2F2) from degradation by the PAXT–exosome complex and thus drives tumor progression [126]. This finding suggests that non-histone lactylation can alter the physicochemical properties of a protein (e.g., phase separation) or interactions to decode the signal, offering a new perspective on lactylation readout mechanisms.
In conclusion, the dynamic regulation of lactation modification constitutes a sophisticated signaling network that relies on the synergistic action of multiple enzymes and effector proteins. To systematically delineate the currently identified key regulatory factors, we have summarized the specific members of the aforementioned “Writers,” “Erasers,” and “Readers,” the target molecules, and functional mechanisms under various physiologic and pathologic conditions in the table below for reference and comparison (Table 1).
Table 1 Summary of Writer, Eraser, and Reader Functions Related to Lactylation Modification
| Type | Protein Name | Function | References |
|---|---|---|---|
| Writer | p300 (CBP/p300 HAT family) | The first identified histone lactylation writer. Catalyzing H3K18la is crucial for the malignant phenotype of pancreatic ductal adenocarcinoma; H3K181a forms a complex with BRG1 and HDAC3 in liver metastasis of colorectal cancer to co-regulate H3K18la. | [17, 108–110] |
| Writer | HDAC1/2/3 | Although generally regarded as a deacylase, HDAC1/2/3 can catalyze lactoylation in reverse under specific conditions, acting as a driver of intracellular lactoylation. | [40] |
| Writer | AARS1 | In esophageal squamous cell carcinoma, as a specific writer of K23 lactylation on RNA processing factor NUDT21. | [112] |
| Writer | KAT8 (Acetyltransferase) | KAT8 acts as a “pan-lacticase writer” in colorectal cancer, catalyzing the lactylation of various substrates, including eEF1A2 (at the K408 site), and promoting translation elongation. | [95] |
| Eraser | HDAC1/2/3 (Class I HDACs) | HDAC1/2/3 is an effective lactate dehydrogenase in mammalian cells. | [113, 114] |
| Eraser | HDAC2 | HDAC2 acts as a specific eraser of H3K9la during angiogenesis (forming a feedback loop); HDAC2 has been identified as a potential histone delactylase in pancreatic ductal adenocarcinoma. | [109, 115] |
| Eraser | HDAC3 | Removing H4K12la in macrophages to regulate downstream TGF-β signaling; HDAC3 is reported to be the delactoylase of NBS1. | [111, 116] |
| Eraser | HDAC8 | PRMT1 is a specific de-lactoylase; HDAC8 is downregulated by hypoxia during breast cancer metastasis, leading to increased lactoylation of PRMT1 and driving metastasis. | [117] |
| Eraser | Sirtuins (SIRT1–3, SIRT6) | Exhibit delactylase activity with distinct catalytic efficiencies and substrate specificities. | [21, 114] |
| Eraser | SIRT1 | Remove the lactylation of histone H3K18la; delactate the α-myosin heavy chain (K1897) in the heart to regulate its interaction with myosin. | [118–120] |
| Eraser | SIRT3 | The main binder and specific eraser of H3K9la (inhibiting transcription in esophageal squamous cell carcinoma); also shows strong activity against H4K16la. | [114, 121] |
| Eraser | SIRT6 | The key deacetylase enzyme can remove various histone marks related to metabolism, including lactylation. | [122] |
| Reader | Brg1 (SWI/SNF complex subunit) | The reader of H3K18la. It is specifically recruited by H3K18la during iPSC reprogramming and promotes mesenchymal-epithelial transition (MET). | [123] |
| Reader | DPF2 | The specific binding partner of H3K14la in cervical cancer cells; DPF2 co-accumulates with H3K14la at the promoters of oncogenes to drive transcription. | [124] |
| Reader | TRIM33 (Bromodomain protein) | Bind lactylated histone peptides with sub-micromolar affinity. Read the lactylation signals in macrophages to downregulate late inflammatory genes and regulate polarization. | [125] |
| Reader | YTHDC1 (m6A reader) | By undergoing auto-lactylation at its K82 site, YTHDC1 “reads” the signal. Lactylation enhances its phase separation ability in the cell nucleus, thereby protecting the oncogene transcripts. | [126] |
Histone lactylation
Histone lactylation was first reported in Nature in 2019 by Zhang et al. as a novel lysine PTM that overturned the traditional notion of lactate as mere metabolic “waste” [17]. This modification directly encodes the metabolic signal of intracellular lactate accumulation onto chromatin by covalently attaching lactyl groups to histone lysines, thereby finely coupling metabolic reprogramming with epigenetic regulation [17, 127]. Like classical histone acetylation and methylation, histone lactylation is reversible and dynamic, engaging a modular “writer–eraser–reader” system across diverse physiologic and pathologic contexts, and serves as a crucial bridge linking cellular energetic state to remodeling of gene expression programs [17, 127].
Molecular mechanism: dynamic balance of “writing–erasing”
Histone lactylation levels depend on intracellular lactate that are typically derived from glycolysis or the Warburg effect [17, 128]. Formation relies on writers that catalyze transfer of lactyl groups from lactyl-CoA to specific histone lysines [128]. Studies have suggested that EP300 of the p300/CBP family can act as a writer. A GTP-specific succinyl-CoA synthetase may function as a nuclear lactyl-CoA synthase that cooperates with p300 to promote histone lactylation [129]. As with other dynamic PTMs, erasers exist (e.g., HDAC1–3 may remove lactylation) [130]. HDAC2 has been further identified as a key eraser that downregulates sites, such as H3K18la, in pancreatic cancer cells, thereby partially suppressing oncogenic transcriptional programs [109]. The dynamic balance between writing and erasing precisely controls lactylation levels and function. Chronic disequilibrium drives epigenetic reprogramming in cancer, and cardiovascular and inflammatory diseases.
Core sites
Twenty-eight lactylation sites have been identified on core histones in human and murine cells [17]. Among the the lactylation sites, H3K18la and H3K9la are the most thoroughly studied. H3K18la is the best-characterized functional mark and serves as a persistent epigenetic mark of innate immune memory that can be detected up to 90 days post-vaccination [131]. H3K18la primes transcription of key functional genes in CD8+ T cells [132]. H3K18la drives tumor growth, immune evasion, and therapy resistance by activating oncogenes, such as ACAT2 [133], TTK [109], BUB1B [109], and CD47 in multiple cancers (e.g., pancreatic cancer and glioblastoma) [134]. H3K9la also has a critical role in CD8+ T-cell differentiation [132]. H3K9la and H3K18la cooperate in epigenetic reprogramming to maintain stemness in pancreatic cancer stem cells [135]. H3K9la and H3K14la promote expression of osteogenic genes (e.g., Runx2 and BMP2) in calcific aortic valve disease, driving valvular calcification [136]. In addition, bacterially induced H3K9la promotes Arg1, which is linked to wound healing and tissue homeostasis, facilitating a shift from pro-inflammatory to reparative states in late-phase M1 macrophage activation [17].
Biological functions
Lactate-driven histone lactylation (e.g., H3K18la and H3K9la) is enriched at promoters and enhancers at the global level, increasing chromatin accessibility, recruiting transcription factors and coactivators, and directly promoting target gene transcription [17, 131, 137]. The lactate-driven histone lactylation functions are broad, as below.
Physiology (development and cell fate). Histone lactylation in glycolytically active cells, such as neural crest, and marks enhancers of key developmental genes and helps deploy regulatory networks during embryogenesis [23]. Lactate-driven histone lactylation reprograms immune-cell function in immunity and inflammation. Elevated H3K18la directly activates Arg1, Lrg1, VEGF-α, and IL-10 in late-phase M1 macrophages or after myocardial infarction, which promotes tissue repair, angiogenesis, and anti-inflammatory programs [17, 138]. Fluctuations in lactylation patterns fine-tune immune transcriptomes, balancing pro- and anti-inflammatory pathways in sepsis and other systemic inflammatory states [139].
Pathology. Aberrant lactylation activates harmful gene programs. Histone lactylation is a key epigenetic driver of tumorigenesis, activating oncogenic transcription to promote proliferation, migration, stemness, and therapy resistance. H3K18la forms a positive feedback loop with glycolysis in pancreatic ductal adenocarcinoma, activating mitotic checkpoint regulators (TTK and BUB1B) to worsen malignancy [109]. Hypoxia, via the HIF-1α/LDHA axis, drives lactate accumulation and H3K18la in colorectal cancer, directly activating promoters of stemness genes (OCT4 and CD44) [140]. Histone lactylation also remodels the tumor immune microenvironment by inducing M2 macrophage polarization and upregulating immune checkpoints, which fosters immunosuppression [128]. Pathologic cardiac hypertrophy and pulmonary arterial hypertension correlate well with elevated lactylation, particularly H3K18la, driving pathologic gene programs and aberrant proliferation in the cardiovascular system [141, 142]. Lactylation is implicated in Alzheimer’s disease, depression, neuroinflammation, and glioblastoma (e.g., via an H4K12la/PKM2 feedback loop in AD) [143]. Elevated histone lactylation promotes resistance to ferroptosis via the HIF1A/HMOX1 pathway in endometriosis, which fuels disease progression [144].
Overall, histone lactylation is a pivotal bridge between cellular metabolism and the epigenome, spanning cancer, immune regulation, metabolic reprogramming, tissue development and repair, and neuro/cardiovascular diseases, which offers fresh perspectives and targets to decode metabolism–epigenetics crosstalk and to develop new therapies.
Non-histone lactylation
Non-histone lactylation is a newly recognized regulatory mechanism whereby lactyl groups covalently modify lysine residues on non-histone proteins, thereby influencing protein function and activity [145]. Unlike histone lactylation that primarily regulates gene transcription, non-histone lactylation directly affects cellular behavior by altering protein conformation, molecular interactions, enzymatic activity, and subcellular localization [146]. High-throughput proteomics has confirmed widespread non-histone lactylation, which collectively indicate that non-histone lactylation is pervasive, as follows: 1090 sites across 469 proteins in Salmonella [105]; 681 sites across 379 proteins in human prostate cancer cells [147]; and 724 sites across 451 proteins in normal human lung tissue [148].
Dynamic control by enzymatic and non-enzymatic mechanisms
Non-histone lactylation arises via diverse mechanisms, including enzyme catalysis and non-enzymatic reactions. Two main precursors supply lactyl groups (L-lactyl-CoA and S,D-lactoylglutathione) [105, 127]. The former precursor typically donates lactyl groups in enzymatic reactions, whereas the latter precursor directly transfers lactyl groups to lysine non-enzymatically [127]. Lactate is converted into active donors (e.g., L-lactyl-CoA) and specific writers, such as p300/CBP or newly identified enzymes (e.g., AARS1/2, YiaC), which catalyzes transfer to targets in enzymatic routes [40, 127, 149]. Conversely, non-enzymatic modification occurs via S–D-lactoylglutathione directly reacting with lysines [105, 127]. Lactylation is reversible. Delactylases, including deacetylases and sirtuins (e.g., CobB), remove lactyl groups to dynamically control modification levels [149, 150].
Biological functions
Non-histone lactylation has central roles across physiology and pathology. Non-histone lactylation modifies key metabolic enzymes, transcription factors, and signaling proteins in cancer to reshape metabolic reprogramming, proliferation, invasion, and the immune microenvironment [127, 146, 151–153]. Lactylation participates in macrophage polarization toward anti-inflammatory phenotypes and is closely associated with diseases, including diabetic nephropathy [154], cardiovascular disorders [155], neuropsychiatric diseases [156], and bacterial infections [105]. Future research should delineate specific enzymatic networks, crosstalk with other PTMs, and advance clinical translation as biomarkers and therapeutic targets.
Functions of lactylation
Regulation of inflammation
Lactylation has central regulatory roles in the initiation, amplification, and resolution of inflammation [157, 158]. Lactylation is a core mechanism in innate immunity that shapes macrophage phenotype and inflammatory balance. Macrophages enhance glycolysis and accumulate lactate in pro-inflammatory states, driving histone lactylation that directly promotes transcription of anti-inflammatory and reparative genes. For example, mitochondrial fragmentation elevates lactate and histone lactylation, upregulating Arg1 and promoting a shift toward M2, which is crucial for timely resolution and tissue repair [159]. Lactylation in macrophages similarly induces VEGF-A and IL-10 early after myocardial infarction, fostering cardiac repair. Conversely, aberrant lactylation aggravates injury. Silica-induced glycolytic reprogramming in silicosis models elevates lactate and histone lactylation, activating the NLRP3 inflammasome, triggering pyroptosis, and sustaining lung inflammation [160]. High expression of lactylation-related genes correlates with immune cell infiltration and pro-inflammatory cytokine release in spinal cord injury [161].
Lactylation profoundly modulates adaptive immunity in the tumor microenvironment, especially T-cell function. Lactylation can bidirectionally tune T cells depending on metabolic context. More often, lactylation favors regulatory programs and contributes to multi-faceted immunosuppression aiding tumor escape [162]. Lactate-driven H3K18la transcriptionally activates ACAT2 in pancreatic cancer, forming a positive feedback loop that promotes release of cholesterol-enriched small extracellular vesicles, polarizes tumor-associated macrophages toward immunosuppressive M2, and weakens antitumor immunity [133]. Lactylation is also pivotal in antiviral innate immunity. Lactylation of the RNA m6A demethylase, ALKBH5, is essential to initiate effective responses during DNA herpesvirus and monkeypox infection. Viruses enhance interaction of the acetyltransferase, ESCO2, with ALKBH5 to promote lactylation. Lactylated ALKBH5 binds and demethylates IFN-β mRNA, boosting IFN-β mRNA production and inhibiting viral replication [163].
Lactylation underlies neutrophil-driven inflammation in myocardial ischemia–reperfusion injury in cardiovascular disease. S100a9 lactylation at K26 enables nuclear entry and coactivator functions to drive transcription of migration genes and neutrophil recruitment. Lactylated S100a9 is released and disrupts mitochondrial function during NETosis, causing cardiomyocyte death [164].
In sum, lactylation promotes inflammatory resolution and tissue repair under physiologic conditions, yet in chronic diseases (cancer, autoimmunity, fibrotic disorders, and neurodegeneration) and acute injuries, aberrant lactylation sustains pathologic inflammation by modulating immune-cell function, pyroptosis, and fibrosis [158, 165].
Angiogenesis
In addition to inflammation, lactylation has important roles in angiogenesis. Lactylation forms a self-amplifying positive-feedback circuit in endothelial cells that directly drives angiogenic programs. VEGF stimulation specifically upregulates H3K9la, which then accumulates at promoters of pro-angiogenic genes and activates the transcription. Notably, high H3K9la suppresses its eraser, HDAC2, forming an H3K9la/HDAC2 positive feedback loop that amplifies VEGF signaling [115]. Similarly, the transmembrane protein, SEMA6A, undergoes liquid–liquid phase separation via its C-terminal intrinsically disordered region to form condensates that recruit RHOA and p300. This finding promotes p300 phosphorylation and lactyltransferase activity. The process catalyzes histone lactylation, which in turn enhances glycolysis and lactate generation, constituting a SEMA6A-centered loop that couples phase separation, metabolism, and epigenetic modification to sustain pathologic angiogenesis [166]. DNMT3A facilitates nuclear entry of lactate and catalyzes lactylation of HIF-1α under hypoxic conditions, markedly upregulating VEGF-A to drive angiogenesis [167].
Lactylation also regulates angiogenesis in tumor and retinal pathologies, often indirectly via RNA modification or immune cell function. Lactate-mediated histone lactylation promotes transcription of ST2 in tumor-associated endothelial cells, enhancing responsiveness to the angiogenic cytokine IL-33 in melanoma [168]. Histone lactylation upregulates the RNA methyltransferase, NSUN2, which increases m5C on AKAP2 mRNA to activate the PKA–VEGFR2 pathway and drive disease in choroidal neovascularization [169]. Enhanced monocyte glycolysis in hyperglycemia and H4K8la increase the content of the pro-angiogenic factor, periostin, in exosomes. These exosomes stabilize HIF-1α and promote retinal neovascularization [170]. Microglial YY1 is lactylated by p300 in hypoxic retinas and directly activates FGF2 transcription to stimulate angiogenesis [171].
Metabolic reprogramming
Metabolic reprogramming is a fundamental process by which cells systematically reshape gene expression, signaling pathways, and enzyme activities in response to internal/external stresses to actively adjust nutrient uptake/utilization and energy production [172, 173]. Cells reprogram metabolism with a predominant enhancement of glycolysis in tumors, inflammation, and hypoxia, and even with sufficient oxygen, cells favor rapid glycolytic ATP production and accumulate lactate, which provides substrate for lactylation [174, 175]. Specifically, activation of HIF-1α and c-Myc upregulates key glycolytic enzymes (HK2, PKM2, and LDHA) in cancer, driving this process [174]. Accumulated lactate, which is catalyzed by CBP/p300, modifies histone lysines to alter chromatin and directly promote oncogenic transcription [137, 174, 175]. Lactylation also targets non-histone proteins, such as p53 and PD-L1, regulating stability or function to support tumor growth, metastasis, and immune escape [176]. This modification suppresses antitumor immunity by upregulating oncogenic pathways, inducing M2 macrophage polarization and causing T-cell dysfunction [174].
Lactylation is the core link between metabolic reprogramming and immune cell function. Activated macrophages generate lactate via glycolysis. Lactylation promotes transcription of chemokines, such as CXCL16, recruiting cytotoxic T cells and exacerbating tissue damage in spinal cord injury models [177]. Lactate accumulation and lactylation can impair immune cell function (e.g., inducing M2 polarization, T-cell dysfunction, and dampening antiviral signaling) in tumor microenvironments or viral infections, thereby shaping immunosuppression and enabling tumor or viral immune escape [178, 179].
Lactylation directly tunes metabolic pathways and cellular homeostasis by modifying key metabolic enzymes and structural proteins at the level of organelle function and cell fate. Dexmedetomidine modulates metabolic reprogramming to lower lactate, thereby inhibiting lactylation of malate dehydrogenase 2, mitigating mitochondrial dysfunction and ferroptosis, and improving myocardial ischemia–reperfusion injury [180], which illustrates the therapeutic potential of correcting pathologic metabolic imbalance by targeting specific lactylation events. Another study showed that the autophagy kinase, ULK1, phosphorylates and activates LDHA under nutrient deprivation to boost lactate production. Lactate then lactylates Vps34, enhancing the lipid kinase activity, promoting autophagic flux and endosome–lysosome trafficking, and integrating two central processes (autophagy and glycolysis) [181]. Dysfunction of DAPK2 induces lactylation of the inner-membrane protein Mic60 at the mitochondrial level, altering cristae architecture, activating mitochondrial metabolism, and ultimately promoting EGFR-TKI resistance and metastasis in lung cancer [182]. These findings indicated that lactylation can precisely target intracellular nodes to reprogram energy metabolism, autophagy, and cell death.
In summary, lactylation and metabolic reprogramming are inseparably intertwined in a bidirectional relationship. Metabolic reprogramming, especially a shift toward aerobic glycolysis, is the fundamental driver of lactate accumulation and lactylation [174, 175]. Transcription factors, such as HIF-1α and c-Myc, upregulate HK2, PKM2, and LDHA, accelerating lactate production and supplying abundant substrate for lactylation [174]. In contrast, lactylation profoundly reshapes cellular metabolism by modifying metabolic enzymes (e.g., ALDOA) or regulating transcription of metabolism-related genes, lactylation sustains PI3K–AKT–mTOR and Wnt/β-catenin survival pathways, consolidating and amplifying the glycolytic phenotype and achieving persistent metabolic reprogramming [183].
Other functions
In addition to established roles in inflammation regulation, angiogenesis, and metabolic reprogramming, an increasing body of evidence indicates that lactylation serves as a pivotal “metabolic-epigenetic hub,” broadly linking cellular metabolic states to cell fate determination, genomic stability, and tissue remodeling [17, 184, 185].
Glycolysis-driven histone lactylation specifically marks the chromatin of highly glycolytic embryonic tissues, such as the neural crest and presomitic mesoderm, in embryonic development and stem-cell biology, thereby opening critical enhancers and activating the neural-crest gene-regulatory network. This process effectively “writes” local metabolic states into developmental programs [17, 23]. Recent developmental studies have further shown that inhibition of lactate production or disruption of histone lactylation deposition results in down-regulation of neural-crest genes, impaired cell migration, and aberrant lineage specification, suggesting that lactylation constitutes an essential epigenetic mark required for early morphogenesis and organogenesis [23]. Enhanced glycolytic flux induces lactylation of the transcription factor, Esrrb, in embryonic stem cells, which strengthens the promoter-binding affinity, maintains the pluripotency transcriptional network, and promotes differentiation toward extra-embryonic endoderm-like stem cells, which highlights a fine regulatory role of non-histone lactylation in cell-fate decisions [186]. Studies involving bone metabolism have demonstrated that lactylation modulates gene expression in osteoblasts and osteoclasts and influences bone-immune crosstalk mediated by macrophages and T cells. Lactylation thus has key roles in bone formation, resorption, and mass maintenance with aberrant lactylation observed in disorders, such as osteoporosis, rheumatoid arthritis, and periodontitis [187]. Systematic analyses further revealed that the interplay among lactylation, metabolism, and immunity within bone tissue governs bone-remodeling homeostasis and provides a conceptual framework for therapeutic strategies that target lactylation to enhance bone regeneration, repair skeletal defects, and treat metabolic bone diseases [184, 187].
Lactylation directly participates in the DNA-damage response and homologous-recombination repair at the level of genomic stability. Chen et al. demonstrated that lactylation of MRE11 at specific residues promotes stable assembly of the MRN complex and accumulation at double-strand-break sites, thereby enhancing homologous-recombination efficiency and cellular tolerance to genotoxic stress [188]. Subsequent work by the same group showed that NBS1 K388 lactylation is indispensable for MRN complex functionality and for repairing chemotherapy-induced DNA damage. MRN complex upregulation markedly increases homologous-recombination capacity and contributes to chemoresistance in tumor cells [111]. Lactylation has emerged as a crucial bridge connecting metabolism, neuronal activity, and behavioral phenotypes within the nervous system. Neuronal excitation, electroconvulsive stimulation, or social defeat stress synchronously elevate brain lactate levels and protein lactylation in the mouse prefrontal cortex, accompanied by increased c-Fos expression, reduced social interaction, and heightened anxiety-like behavior, indicating that activity-induced lactylation participates in the epigenetic regulation of emotion and stress responses [189]. Moreover, metabolic-epigenetic studies in the brain revealed that exogenous lactate under hypoxic conditions induces H3K9 lactylation in neural stem cells, upregulates the transcription factor, SnoN, promotes neuronal differentiation, and thereby enhances adult neurogenesis and functional recovery following ischemic injury [190]. In contrast, lactate accumulation in senescent microglia causes marked H3K18 lactylation in natural aging and Alzheimer’s disease models, activating the NF-κB pathway and driving a pro-inflammatory senescence-associated secretory phenotype, which accelerates brain aging and neurodegeneration [191]. Astrocytic LRP1 attenuates glycolysis and suppresses ARF1 lactylation in ischemic-stroke models, thereby facilitating intercellular mitochondrial transfer to damaged neurons and alleviating ischemia–reperfusion injury [192].
Taken together, these findings demonstrate that lactylation exerts multilayered and multifaceted functions across biological systems, including development, bone remodeling, genome maintenance, and neural homeostasis. This reinforces the concept of lactylation as a central integrator linking metabolic cues to cellular function and highlights the need for future therapeutic designs to consider context-dependent and organ-specific effects within the broader landscape of metabolism-epigenetics interactions.
Lactylation modification in PF
Pathologic localization of lactylation in PF
Lactylation, as an emerging form of epigenetic regulation, is increasingly recognized as a key molecular link between glycolytic reprogramming and profibrotic phenotypes in the complex progression of PF. Lactylation reflects the abnormal accumulation of lactate in lesion-resident cells under conditions, such as hypoxia, inflammation, and metabolic stress. In contrast, lactate can mediate histone and non-histone lactylation to finely regulate the expression of genes involved in critical processes, including the EMT, macrophage polarization, fibroblast activation, and endothelial–mesenchymal transition (EndMT). In this way, lactate provides a unified molecular framework for understanding PF as a multicellular pathologic process [17]. Therefore, lactylation is not an isolated event but is embedded within a continuous pathologic axis of “metabolic reprogramming–epigenetic remodeling–cell fate transition.”
Notably, PF exhibits a marked age dependency. Senescent cells are widely distributed in fibrotic lung tissues and are considered an important pathologic basis driving disease progression [193, 194]. Mechanistically, senescent cells are characterized not only by irreversible growth arrest but also by profound metabolic reprogramming. Previous studies have shown that certain senescent cells can upregulate pyruvate dehydrogenase kinase 4, thereby inhibiting the entry of pyruvate into mitochondrial oxidative metabolism. This shift enhances aerobic glycolysis and promotes lactate production, leading to a “hypercatabolic” state that is characterized by elevated glycolytic activity, increased lactate accumulation, and sustained oxidative metabolism. This metabolic phenotype not only reshapes the local acidic microenvironment but also amplifies the senescence-associated secretory phenotype (SASP) and the paracrine pathogenic effects [34].
Accumulating evidence from multiple organ systems suggests that the lactate/lactylation axis fundamentally acts as a mediator that converts metabolic abnormalities in senescent cells into persistent inflammatory responses and tissue remodeling programs. By integrating findings from PF research, it is reasonable to infer that a similar mechanism exists in fibrotic lung tissue. First, significant abnormalities in lactate metabolism have been demonstrated in lung tissues and AEC2s from patients with idiopathic PF, indicating a metabolic basis for sustained lactate accumulation within lesions [11, 67]. Second, elevated lactate levels in fibrotic lungs not only activate latent TGF-β by lowering local pH, thereby promoting myofibroblast differentiation, but also serve as an epigenetic substrate to induce histone lactylation in macrophages and enhancing the pro-fibrotic secretory phenotype [11, 82]. In addition, lactate accumulation mediated by the Akt2–PDK1 signaling pathway has been shown to contribute to the progression of PF [195].
Taken together, senescent epithelial cells, fibroblasts, and immune cells in fibrotic lung tissue may collectively establish a continuous regulatory axis in which senescence-associated metabolic reprogramming drives lactate accumulation, which in turn induces histone and non-histone lactylation, ultimately amplifying SASP and the transcription of pro-fibrotic genes. This coordinated process promotes aberrant tissue repair, pathologic intercellular communication, and ultimately irreversible fibrotic progression.
Lactylation roles in different cell types
To further elucidate the pathogenic significance of lactylation in PF, it is necessary to systematically examine lactylation functions across key effector cell types, including AECs, macrophages, fibroblasts, and endothelial cells.
AECs
Environmental exposures, especially respirable particulate matter, are important etiologies of PF. Evidence indicates that the pathogenic effects critically depend on a signaling axis that serially links inflammatory activation, glycolytic reprogramming, lactate accumulation, and histone lactylation, culminating in EMT of alveolar/airway epithelial cells as the core phenotypic output. Pathogenesis begins with activation of the NLRP3 inflammasome and increased IL-1β secretion in silica exposure models [72]. Subsequently, IL-1β/IL-1R signaling markedly upregulates the key glycolytic regulator, PFKFB3, elevating glycolytic flux and driving robust lactate production [72]. Accumulated lactate then serves as substrate to catalyze histone H3K18 lactylation. Mechanistically, H3K18la is specifically enriched at the promoter of SIX1, a core EMT transcription factor, thereby markedly enhancing transcription and ultimately driving phenotypic conversion of AECs, an early, critical event in PF formation [72].
PM2.5 induces glycolytic reprogramming and elevated histone lactylation in macrophages, the inflammatory factors of which further activate TGF-β/Smad signaling and induce EMT in AECs [24]. More mechanistically, macrophage H3K18la upregulation driven by lactate accumulation suppresses transcription of the E3 ubiquitin ligase, CHIP [79]. Reduced CHIP expression weakens degradation of TGF-β1, increasing stability and activity. Thus, PM2.5 establishes a pathogenic positive-feedback loop (lactate–H3K18la–CHIP–TGF-β1) that promotes fibrotic progression.
Macrophages
As central immune regulators in the PF microenvironment, macrophages exhibit both cell-autonomous and non-autonomous regulation by lactylation. Stimuli, such as silica, directly induce glycolytic reprogramming in alveolar macrophages, increasing lactate production and lactylation in the cell-autonomous mode [160]. Lactate and protein lactylation synergistically promote assembly of the NLRP3 inflammasome and activation of caspase-1, amplifying inflammation and promoting pyroptosis, with abundant release of IL-1β and other mediators (e.g., IL-18, TNF-α, IL-6, and chemokines)—laying a chronic inflammatory foundation in early fibrosis [160]. In the non-autonomous mode, activated pulmonary myofibroblasts function as “metabolic engines,” continuously exporting lactate to the microenvironment through enhanced glycolysis [82]. Surrounding macrophages take up this exogenous lactate, further elevating intracellular lactate and histone lactylation. For example, p300-mediated histone lactylation upregulates transcription of a series of profibrotic genes, polarizing macrophages toward profibrotic states and prompting secretion of factors that activate fibroblasts and drive ECM deposition, thus building a metabolism–epigenetics circuit that sustains fibrosis [82]. Macrophages serve as a key hub linking immune inflammation in PM2.5 models to structural remodeling through the H3K18la–CHIP–TGF-β1 axis and secretion of TGF-β1 and other factors (e.g., PDGF, CTGF, IL-6, and MCP-1) [24, 79].
Fibroblasts
Lactylation regulates activation through multilayered, networked mechanisms in fibroblasts, the effector cells of PF. At the transcription-factor level, TGF-β–induced glycolysis and lactate accumulation can cause specific lactylation of Snail1, potentially directly tuning the pro-fibrotic function. The natural compound, wogonoside, reduces lactate generation and Snail1 lactylation by modulating metabolic enzymes, thereby suppressing fibroblast activation [196].
A more exquisite cascade has been delineated at the epigenetic crosstalk level. H3K9 lactylation enhances transcription of the m6A methyltransferase METTL3 and METTL3 upregulation in turn installs m6A on specific long non-coding RNAs via YTHDC1-mediated phase separation and nuclear export. These RNAs ultimately stabilize translation of pro-fibrotic targets in the cytoplasm, fully activating fibroblasts [197]. This signaling axis sequentially undergoes histone lactylation regulation, RNA m6A modification reprogramming, and enhanced RNA nuclear export, systematically expanding our understanding of the complexity of epigenetic regulatory networks.
Endothelial cells
The spectrum of lactylation also extends to the vasculature. Multi-omics in IPF patients showed that globally elevated lactylation in lung tissue correlates with disease severity and identified IGFBP7 and CCT2 as lactylation-driven key molecules in endothelial cells [198]. Mechanistically, TGF-β induces metabolic reprogramming in the endothelium. The consequent high-lactate milieu drives expression of these molecules and promotes EndMT, providing new explanations for endothelial contributions and perivascular fibrosis in PF [198]. Figure 8 provides a schematic overview of lactylation-driven mechanisms in PF, illustrating how lactate accumulation and lactylation influence key processes in AECs, macrophages, fibroblasts, and endothelial cells, ultimately contributing to the progression of PF.
Figure 8 Schematic diagram of lactic acidosis-driven mechanisms of pulmonary fibrosis initiation and progression. Various environmental and inflammatory stimuli induce glycolytic reprogramming in alveolar epithelial cells, macrophages, fibroblasts, and endothelial cells, leading to excessive lactate production. Local lactate accumulation further drives histone and non-histone acylation, remodeling the expression programs of key genes involved in EMT, macrophage polarization, fibroblast activation, and EndMT. Different cells amplify fibrotic signals through lactate and multiple cytokines, forming a metabolic-epigenetic network centered on acylation, ultimately resulting in excessive matrix deposition and progression of pulmonary fibrosis.
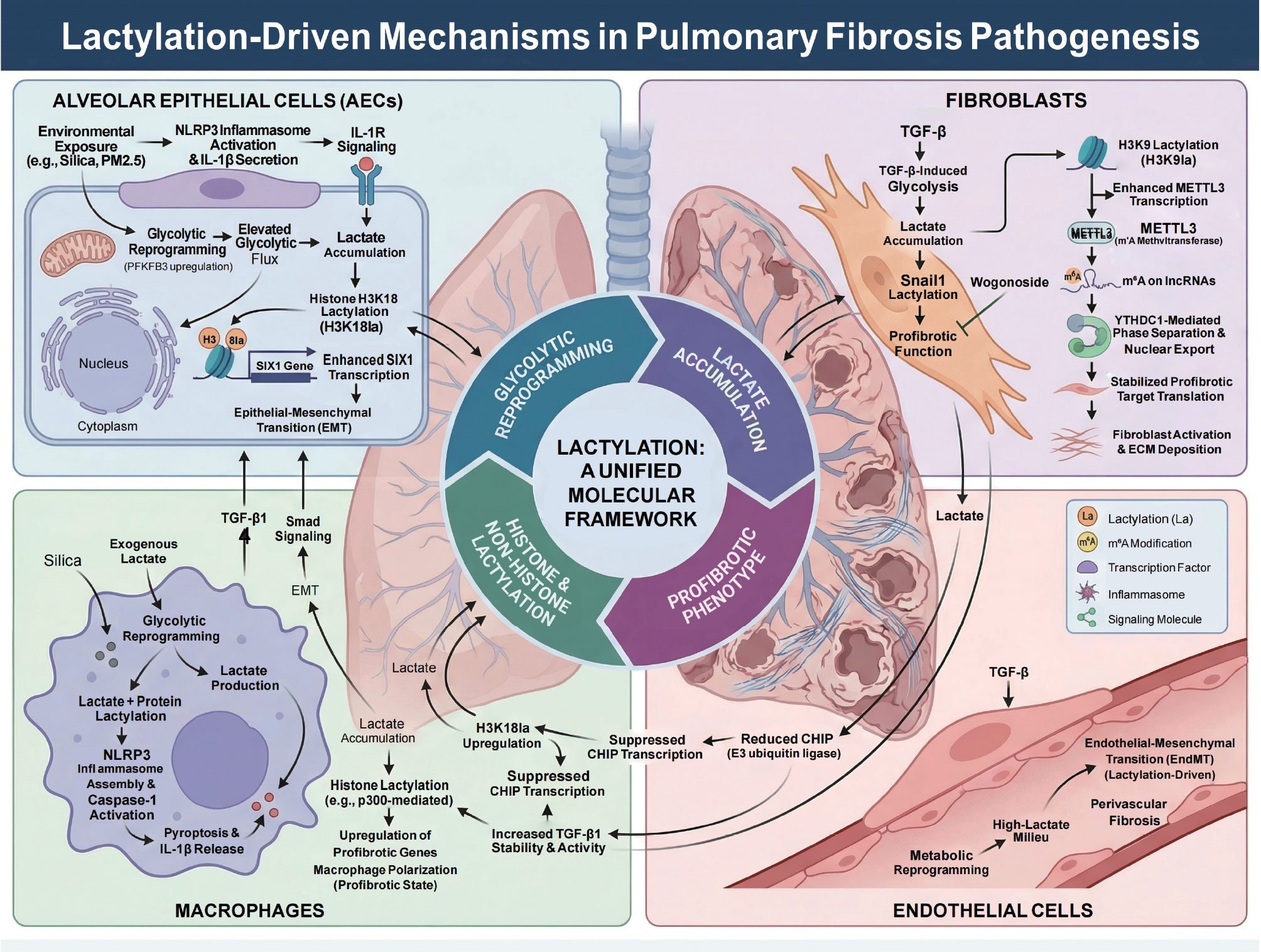
Crosstalk of lactylation with other modifications in PF
Lactylation is not isolated in the intricate pathologic network of PF. Rather, lactylation is deeply embedded in a tightly coupled regulatory system spanning epigenetics–epitranscriptomics–post-translational control. Lactylation shares lysine sites and components of writer/eraser machineries with canonical modifications, such as acetylation, methylation, and β-hydroxybutyrylation, and interlaces with the canonical modifications along metabolic–signaling axes. Thus, lactylation is best viewed in PF as a second signaling layer superimposed on a pre-existing map of chromatin marks, encoding the dynamics of glycolytic reprogramming, rather than a simple replacement [17, 26]. Accordingly, PF does not merely reflect elevated lactylation but a composite epigenetic state shaped by interactions among multiple PTMs.
Histone-level interplay
Lactylation and classical marks (e.g., acetylation) exhibit conspicuous site competition and functional synergy at the histone level. For example, H3K18 can be acetylated, lactylated, or β-hydroxybutyrylated [17, 113]. p300/CBP functions as a shared writer for H3K18ac, and when lactyl-CoA is abundant, H3K18la. HDAC1–3 are shared erasers with deacetylase and delactylase activities [113]. Hence, in the PF milieu of heightened glycolysis and lactate, lactylation can partially displace acetylation/propionylation at specific lysines, reshaping local chromatin “mark cocktails” and rewriting enhancer/promoter states [17, 26]. While PF studies often focus on single marks (e.g., H3K18la and H3K9la), cross-mark dynamics at the same site likely determine priority activation or silencing of specific genes during fibrosis, an area ripe for investigation using quantitative proteomics and multiplex ChIP-seq.
A cascade with RNA m6A modification
Crosstalk between lactylation and RNA m6A modification has been increasingly delineated across disease models and analogously observed in lung injury/fibrosis. Lactate accumulation elevates H3K18la and upregulates METTL3 transcription in tumor-infiltrating myeloid cells. Concurrently, METTL3 undergoes lactylation at a zinc-finger domain, enhancing recognition/catalysis on JAK1 mRNA, thereby activating the JAK1–STAT3 axis and driving immunosuppression [199]. Lactate activates METTL3 via p300-mediated H3K18la in sepsis-associated acute lung injury, which installs m6A on ACSL4 mRNA to promote ferroptosis, suggesting a similar H3K18la–METTL3–m6A axis in alveolar epithelium [200]. H3K9la upregulates METTL3 in PF, which then modifies specific lncRNAs with m6A through YTHDC1-mediated phase separation and nuclear export. Multiple pro-fibrotic translation pathways are stabilized, achieving multistage amplification from “lactate–histone lactylation” to “RNA epigenetics” to “protein synthesis” [197]. Moreover, H3K18la elevates YTHDF1 in arsenic-related PF models, modulating downstream NREP m6A and sustaining TGF-β1 signaling, which further supports the generality of the lactylation–m6A axis in fibrosis [71]. Collectively, these findings depict a representative cross-mark cascade, as follows: intracellular lactate first drives histone lactylation, which then upregulates METTL3 or YTH-family readers (or directly lactylates them); the modified/induced METTL3/YTH proteins remodel the m6A landscape; and ultimately, stability and translational efficiency of pro-fibrotic transcripts are enhanced, amplifying fibrotic outputs [71, 197, 199].
Interplay with ubiquitination and protein homeostasis
Interactions between lactylation and ubiquitination/protein homeostasis are also noteworthy. Elevated macrophage lactate and H3K18la do not merely activate canonical TGF-β/Smad signaling in PM2.5-induced PF. Rather, H3K18la enriched at the CHIP E3-ligase promoter suppresses transcription [24, 79]. Because CHIP governs TGF-β1 degradation, downregulation directly impairs ubiquitin-mediated turnover, causing abnormal extracellular accumulation of TGF-β1 [79]. From a cross-PTM perspective, this finding indicates that lactylation can indirectly rewrite the ubiquitination–proteostasis network by repressing specific E3 ligases and transforming transient metabolic signals into persistent buildup of pro-fibrotic factors. Similar lactylation–ubiquitination interactions have been reported in DNA-damage repair and bone remodeling [111], suggesting additional, yet-uncovered nodes in PF that regulate turnover of ECM proteins, receptors, and signaling molecules.
Crosstalk with other modifications
More broadly, lactylation may form complex “combinatorial codes” with methylation, phosphorylation, β-hydroxybutyrylation, and other post-translational modifications (e.g., acetylation, ubiquitination, and sumoylation). Lactylation often occurs at hotspot lysines enriched for other marks (e.g., H3K9 and H3K18 that carry acetylation/methylation) [17]. In contrast, lactylation-driven transcriptional changes can secondarily alter expression of DNA methyltransferases, histone methyltransferases/demethylases, and m6A methyltransferases that potentially establish temporal cascades in which “lactylation precedes methylation/demethylation” [26, 199]. Systematic multi-omics to delineate spatiotemporal dynamics of such crosstalk in PF are lacking, yet evidence from cancer, cardiovascular, and neurodegenerative fields strongly suggests lactylation acts as a network hub rather than an isolated pathway [26, 39, 113].
These cross-modifications present both opportunities and challenges in PF research. Axes, such as metabolism–lactylation–m6A and metabolism–lactylation–ubiquitination, offer multi-tiered targets. One axis could suppress lactate production/transport (e.g., LDHA and MCT1/4), directly modulating writers, like p300, or erasers (HDAC1-3), or co-target downstream effectors, such as METTL3/YTH family or CHIP [17, 113, 199]. Conversely, the coexistence of multiple axes implies that simple single-target interventions may trigger compensatory pathways or even immunosuppression. Thus, interventions must be designed with cell-type and disease-stage precision.
In sum, lactylation in PF is not an isolated “new mark,” but a core nexus spanning metabolic reprogramming, epigenetic remodeling, and protein fate control. Dissecting crosstalk with other epigenetic and post-translational modifications will help re-understand PF at the network level and lay the groundwork for developing metabolism-epigenetics combination strategies with stronger specificity and more controllable toxicity.
Dynamic lactate-lactylation network in PF
Recent studies indicate that lactate metabolism and lactylation are not confined to a single cell type during PF progression but instead form a dynamic intercellular network. Activated fibroblasts, dysfunctional epithelial cells, and polarized macrophages all exhibit enhanced glycolytic activity within the fibrotic microenvironment, leading to excessive lactate production. Lactate, mediated by monocarboxylate transporters, can be continuously exchanged among these cell populations, thereby establishing a “lactate shuttle system” that integrates metabolic activities within the tissue [81, 82, 201].
Importantly, lactate functions not only as a metabolic substrate, but also as a signaling molecule and an epigenetic regulator. Intracellular lactate accumulation promotes protein lactylation, including histone modifications, such as H3K18la [17]. This process enables metabolic signals to be directly encoded into transcriptional programs, thereby regulating key profibrotic pathways, including TGF-β signaling, the EMT, and macrophage polarization [17, 24, 73, 82].
Notably, the lactate–lactylation axis establishes multiple positive feedback loops within the fibrotic microenvironment. Lactate-induced lactylation can enhance the expression of certain pro-fibrotic genes and may synergize with glycolytic reprogramming, forming an amplification circuit [11, 81, 82]. Similarly, lactate-mediated macrophage activation and epithelial–fibroblast interactions further amplify cytokine release and ECM deposition, thereby reinforcing the fibrotic niche [81, 82, 202].
Collectively, this dynamic network constitutes a mechanistic framework in which local metabolic stress is progressively amplified and stabilized through epigenetic reprogramming. In this context, lactylation may serve as a critical molecular bridge that converts transient metabolic alterations into sustained transcriptional and phenotypic changes, ultimately driving the irreversible progression of organ fibrosis.
Research limitations, technical perspectives, and therapeutic translation
Research limitations and technical perspectives
In recent years studies on lactate metabolism and lactylation in PF have increased significantly. However, most existing evidence remains largely correlative and true “site-specific causal” evidence has yet to be established. Current studies mainly rely on metabolomics, transcriptomics, lactylation proteomics, ChIP-qPCR, CUT&Tag, immunoblotting, and tissue colocalization analyses to demonstrate that lactate accumulation and elevated global lactylation levels occur in parallel with the EMT, fibroblast activation, pro-fibrotic macrophage polarization, and ECM deposition [17, 35].
For example, increased lactate levels have been demonstrated in lung tissues of IPF patients, promoting TGF-β activation and myofibroblast differentiation [11]. Persistent glycolytic reprogramming has been identified in lung fibroblasts/myofibroblasts [10]. Lactate derived from myofibroblasts can induce histone lactylation in macrophages and enhance the pro-fibrotic secretory phenotype [82]. AECs also exhibit abnormal lactate metabolism [67] and increased H3K18 lactylation (H3K18la). Activation of downstream fibrotic transcriptional programs has been observed in arsenic or silica exposure models [71, 72]. In addition, GLP-1R agonists or glycolysis inhibition strategies can reduce fibrosis accompanied by decreased lactylation levels [87]. Macrophage H3K18la amplifies fibrosis by suppressing CHIP expression and stabilizing TGF-β1 in PM2.5 exposure models [79]. Taken together, these findings support the overall framework of a “lactate shuttle-lactylation-pro-fibrotic transcription” axis, although most conclusions are still at the level of an association.
A major bottleneck lies in the fact that most interventions target upstream metabolic or enzymatic nodes, such as LDHA, MCT1/4, PFKFB3, p300, or HDAC1-3 [113]. While these approaches alter lactylation levels, the approaches also affect glycolytic flux, redox balance, intracellular pH, and other lysine acylations (e.g., acetylation), making it difficult to determine whether specific lactylation sites directly drive fibrotic phenotypes. Thus, the key question is no longer whether lactylation is involved but rather which specific sites, in which cell types, and at which disease stages, causally determine fibrosis initiation and irreversibility.
Notably, other research fields have already provided site-resolved causal evidence. For example, lactylation of MRE11 at K673 promotes DNA end resection and homologous recombination repair [188], while lactylation of NBS1 at K388 directly regulates MRN complex assembly and DNA repair efficiency [111]. These studies demonstrated that lactylation is not merely a biomarker but a functional regulatory event. In contrast, PF research still lacks similarly robust “site-specific necessity” evidence, highlighting the need to shift from global observations to precise, cell- and stage-specific causal analyses.
From a methodologic perspective, gene-editing technologies represent a key breakthrough direction. Base editing can be used to substitute lysine with non-lactylatable residues (e.g., arginine) for lysine residues encoded by AAA/AAG without introducing double-strand breaks, thereby generating endogenous site-blocking models [203, 204]. Prime editing can achieve precise multi-base modifications for more complex edits [205]. These strategies can be applied in AECs, macrophages, fibroblasts, organoids, or conditional knock-in mouse models to edit candidate sites (e.g., H3K18, H3K9, or key non-histone sites), combined with transcriptomics, single-cell sequencing, CUT&Tag, ATAC-seq, and phenotypic rescue experiments to establish direct “site–mechanism–phenotype” causal links.
At the same time, advances in chemical biology provide important complementary tools. Current studies rely heavily on pan-lactylation antibodies and endpoint detection, limiting dynamic and site-specific analyses. Bioorthogonal chemical reporters (e.g., YnLac) enable labeling and enrichment of lactylated substrates in live cells [206]. Protein semi-synthesis and expressed protein ligation allow generation of site-specifically modified proteins or nucleosomes [207, 208], while genetic code expansion offers potential for precise incorporation of lactylation marks [209]. Integration of these approaches with quantitative mass spectrometry, live-cell imaging, and gene-editing models will help shift research from correlation to mechanistic dissection.
Overall, future PF research on lactylation should move beyond differential molecular screening toward building a precise, cell-specific, and temporally resolved mechanistic framework [205, 208]. Only when specific lactylation sites are proven to be essential in fibrosis initiation, maintenance, or irreversibility can lactylation truly transition from a “potential biomarker” to a “therapeutic target”.
Despite these findings, clinical translation faces significant challenges. Lactate metabolism and transport systems are broadly involved in normal tissue repair, immune homeostasis, and systemic energy metabolism. For example, lactate promotes endothelial cell migration and angiogenesis and regulates monocyte/macrophage function [210, 211], while MCTs maintain transport of lactate, pyruvate, and ketone bodies and acid–base balance across tissues [212]. Clinical and genetic evidence further highlights the importance. MCT1 deficiency can cause ketoacidosis and the MCT1 inhibitor, AZD3965, has been associated with severe hyperlactatemia [213, 214]. Similarly, targeting lactylation-related enzymes carries off-target risks. CBP/p300 inhibitors may impair platelet production and cause multi-system toxicity [215]. HDAC inhibitors are associated with immunosuppression and thrombocytopenia [216, 217]; SIRT family proteins are broadly involved in mitochondrial metabolism and immune regulation; systemic inhibition may disrupt homeostasis [218, 219]. Therefore, more feasible strategies may involve local delivery, short-term intervention, and cell-specific targeting, or prioritizing relatively specific downstream effectors within fibrotic tissues to improve safety and efficacy (Figure 9).
Figure 9 Conceptual framework of lactate metabolism and lactylation in pulmonary fibrosis, from current limitations to therapeutic translation. The schematic is organized into three panels. (1) Current limitations: Existing studies largely rely on correlative evidence linking lactate accumulation and global lactylation to key fibrotic processes, including epithelial–mesenchymal transition, fibroblast activation, pro-fibrotic macrophage polarization, and extracellular matrix deposition. Interventions targeting upstream metabolic nodes (e.g., LDHA, MCT1/4, PFKFB3, p300, and HDAC1–3) affect glycolytic flux, redox balance, and intracellular pH but site-specific causal mechanisms remain unclear. (2) Future perspectives: Emphasis is placed on identifying cell type– and stage-specific mechanisms using gene-editing technologies (e.g., base editing and prime editing) and chemical biology tools (e.g., bioorthogonal reporters and genetic code expansion) combined with live-cell labeling and multi-omics approaches to establish direct links between lactylation sites and fibrotic phenotypes. (3) Therapeutic translation: Challenges include off-target effects and systemic toxicity of global inhibition. Proposed strategies highlight targeted, localized delivery (e.g., inhalation), and combination therapies integrating anti-fibrotic drugs (pirfenidone and nintedanib) with metabolic/epigenetic modulators. A translational pathway involving target identification, patient stratification, pharmacodynamic monitoring, and validation is outlined to enable safe and effective clinical application.
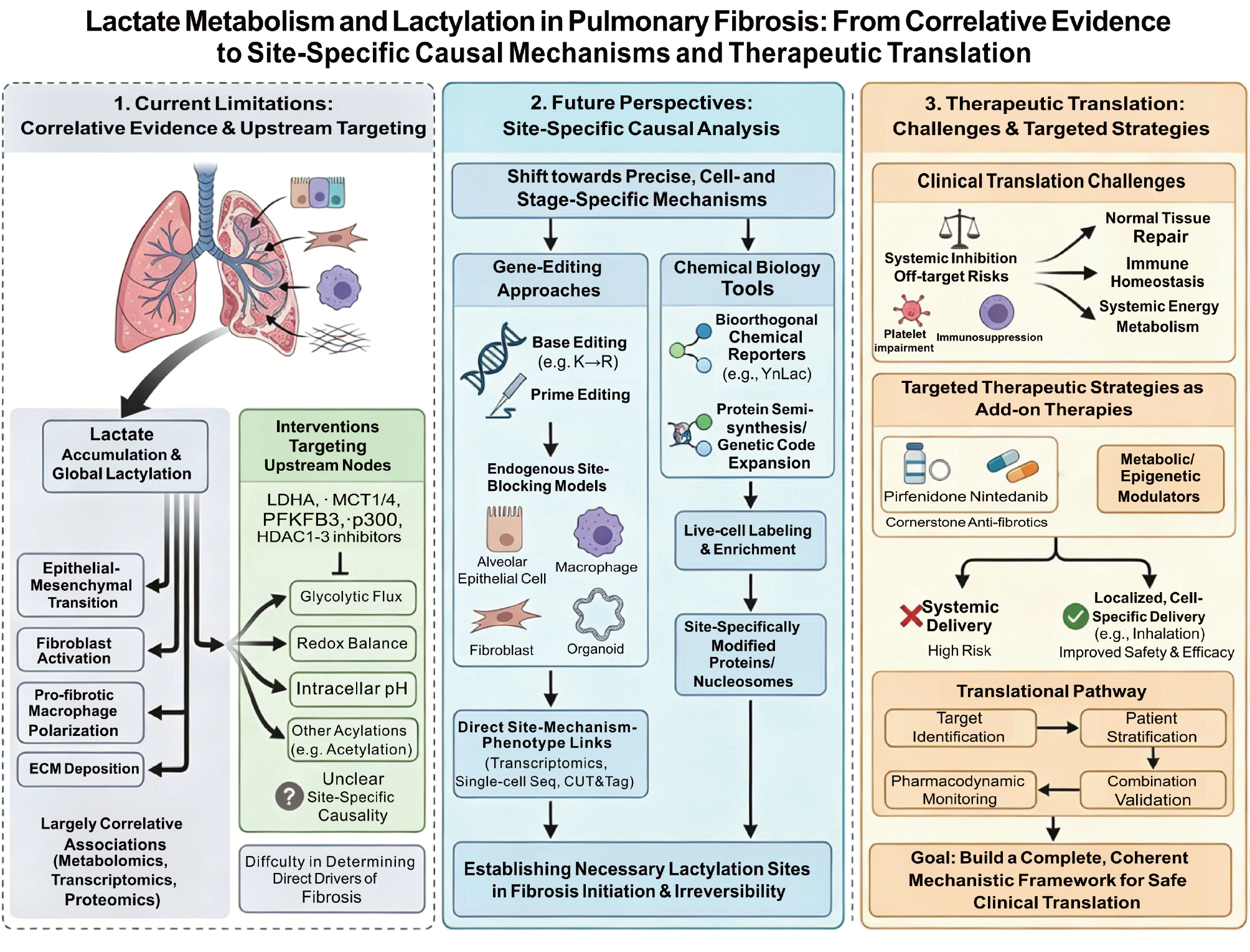
Therapeutic prospects and translational pathways
Currently, pharmacologic treatment of IPF still relies on approved anti-fibrotic drugs as the clinical cornerstone. The ASCEND study demonstrated that pirfenidone slows disease progression, while the INPULSIS study showed that nintedanib significantly reduces the annual decline in forced vital capacity (FVC). The INBUILD and SENSCIS studies further expanded the indications of nintedanib to progressive fibrosing interstitial lung diseases and systemic sclerosis-associated interstitial lung disease [64, 220–222].
Thus, therapeutic strategies targeting the “lactate metabolism-lactylation axis” are more appropriately positioned as add-on therapies rather than replacements for existing anti-fibrotic treatments. Studies also suggest that pirfenidone and nintedanib can be safely combined under some conditions, supporting a combination model of “anti-fibrotic cornerstone drugs + metabolic/epigenetic modulators” [223–225].
Mechanistically, therapeutic development can be divided into three levels: upstream lactate generation and transport (LDHA and MCT1/4); lactylation-modifying enzymes (p300/CBP and HDAC1-3); and downstream effector nodes, including site-specific lactylation and the impact on transcription, RNA modification, or protein homeostasis. Evidence suggests that targeting downstream, cell- and stage-specific nodes may be more effective for early translational breakthroughs.
Given safety concerns, future strategies should prioritize localized, short-term, and reversible interventions. Inhalation-based delivery systems, including inhaled nintedanib and nanoparticle formulations, have demonstrated reduced systemic exposure and improved lung retention, providing a strong methodologic basis for future development.
Translational research may follow four steps: target identification; patient stratification; pharmacodynamic monitoring; and combination validation. Ultimately, successful clinical translation will depend not only on identifying new targets but also on building a complete and coherent translational framework (Figure 9).
Conclusion and perspectives
PF is a progressive disease that is characterized by destruction of alveolar architecture, aberrant fibroblast activation, and persistent ECM deposition. The initiation and progression of PF involve multiple interconnected mechanisms, including impaired epithelial injury repair, remodeling of the immune microenvironment, metabolic abnormalities, and epigenetic regulation. In recent years, with the deepening understanding of metabolic reprogramming, lactate is no longer regarded merely as the end-product of glycolysis but has been redefined as a key molecule with a triple role as a metabolic substrate, an intercellular signaling molecule, and an epigenetic regulatory mediator. In particular, the discovery of lactylation has provided a new theoretical basis for understanding how metabolic abnormalities are translated into stable pathogenic transcriptional programs and has made the “lactate metabolism–lactylation axis” an important entry point for elucidating the pathogenesis of PF.
This review systematically summarizes the generation, transport, and utilization of lactate, and further outlines the molecular basis underlying lactylation and its potential pathogenic significance in different cell types in PF. Current evidence indicates that, AECs, fibroblasts, macrophages, and endothelial cells all undergo varying degrees of glycolytic reprogramming within the fibrotic microenvironment, leading to sustained local lactate accumulation. Elevated lactate can directly promote fibrosis progression by acidifying the microenvironment, facilitating latent TGF-β activation, aggravating endoplasmic reticulum stress, and inducing the EMT. In contrast, lactate can serve as a substrate for lactylation, altering the functional states of both histone and non-histone proteins, thereby regulating gene expression programs associated with inflammatory amplification, cell fate transitions, collagen synthesis, ECM deposition, and pathologic intercellular communication. These findings suggest that lactate not only shapes the metabolic background of PF but may also be deeply involved in maintaining and amplifying pathologic phenotypes.
Furthermore, the significance of lactylation in PF is not limited to changes at a single site or within a single cell type but more likely reflects a dynamic regulatory network spanning multiple cells and multiple levels. Lactylation participates in the activation of EMT-related transcriptional programs in epithelial cells. Lactylation promotes pro-fibrotic polarization and enhances the secretion of factors, such as TGF-β1, in macrophages. Lactylation is coupled with m6A modification, transcription factor activity, and RNA nuclear export in fibroblasts, thereby sustaining fibroblast activation. Lactylation may be involved in the EMT and perivascular fibrosis in endothelial cells. More importantly, lactylation does not occur in isolation but together with acetylation, methylation, ubiquitination, and RNA modifications constitutes a complex network of crosstalk, through which metabolic changes are progressively amplified into stable fibrotic phenotypes. For this reason, lactylation may be regarded as a core bridge linking metabolic reprogramming, epigenetic remodeling, and tissue reconstruction.
Although this field has advanced rapidly, our understanding of lactylation in PF remains at an early stage and several key issues still need to be addressed. First, most current studies remain at the level of correlation, namely “increased lactate–increased acetylation-aggravated fibrotic phenotype,” and lack truly site- and cell-specific causal evidence. Second, the global landscape of histone and non-histone lactylation in PF is incomplete and the spatiotemporal heterogeneity of lactylation across disease stages, cell types, and microenvironmental conditions has not been systematically characterized. Third, the network of lactylation “writers,” “erasers,” and “readers” has not been fully elucidated and the substrate selectivity, upstream–downstream coupling, as well as the competition or cooperation with other post-translational modifications, all require further investigation. In addition, lactate and lactylation also have important physiologic roles in normal tissue repair, maintenance of immune homeostasis, and energy metabolism. This finding means that interventions targeting this pathway must take cell type, disease stage, and tissue specificity fully into account. Otherwise, lactate and lactylation may cause metabolic disturbances, immune suppression, or impaired tissue repair.
Looking ahead, research on lactylation in PF should move gradually from “phenomenon description” to “mechanistic dissection” and “precision translation.” Technologies, such as single-cell transcriptomics, spatial omics, quantitative lactyl-proteomics, multiplex ChIP-seq, CUT&Tag, and metabolic flux tracing, should be used to systematically construct dynamic maps of lactate metabolism and lactylation across different cell types, thereby identifying truly pathogenic key sites and core nodes. In contrast, base editing, prime editing, conditional knock-in models, and organoid systems should be combined to verify the functional necessity of specific lactylation sites in the initiation, maintenance, and irreversibility of fibrosis at the endogenous level, thereby establishing a direct causal chain of “site–cell–disease stage–phenotype.” Greater efforts are needed to investigate the crosstalk between lactylation and m6A modification, ubiquitination, phase separation, mitochondrial homeostasis, and immunometabolic remodeling to reconstruct our understanding of PF pathogenesis from a network perspective.
Targeting the “lactate metabolism–lactylation axis” holds considerable promise At the level of clinical translation. However, a more rational strategy may not be long-term, systemic, broad-spectrum inhibition but rather precise intervention centered on key nodes that are lesion-enriched, cell-specific, and stage-dependent. For example, priority could be given to screening lactylation sites, specific writers/erasers, MCT transport pathways, or downstream effector molecules that are abnormally enriched in fibrotic lung tissue, combined with inhalational delivery, nanocarriers, and local short-course interventions to maximize pulmonary drug accessibility while minimizing systemic side effects. In addition, lactate and lactylation-related molecules may serve as dynamic biomarkers for disease stratification and therapeutic evaluation, helping to identify patient subgroups with more prominent lactate metabolic abnormalities who may be more suitable for metabolic intervention.
Overall, the emergence and expanding understanding of lactylation are driving pulmonary fibrosis research beyond the traditional framework of “inflammation–injury–repair” toward an integrated new paradigm of “metabolism–epigenetics–cell fate.” In the future, the lactate metabolism–lactylation axis is expected not only to deepen our understanding of PF pathogenesis with the continued development of multi-omics technologies, precise disease modeling, and targeted delivery strategies but also provide a solid foundation for establishing new disease stratification systems, identifying druggable targets, and developing more precise combination therapies, ultimately bringing new breakthroughs to the individualized diagnosis and treatment of PF.
Data availability statement
Data will be made available on request.
Author contributions
Fengxu Wang: Writing-original draft, Methodology, Investigation, Data curation. Mengna Jiang and Li Zhu: Writing-original draft, Formal analysis, Data curation. Jiaxin Liu, Jinyu Tang and Xueshan Jin: Writing-original draft, Methodology, Funding acquisition. Hanrui Liu, Ziruo Cheng, Zihan Wang, Rongzhu Liu and Haotian Xu: Writing-original draft, Visualization, Methodology. Bing Han, Demin Cheng and Xinyuan Zhao: Writing-review & editing, Supervision, Project administration, Funding acquisition.
Acknowledgment
The graphical abstract and Figures 1-9 were created using Adobe Illustrator 2022, with the visual style partly inspired by Nano Banana. This work was financially supported by the National Natural Science Foundation of China (82574142, 82504379) and funded by Basic Research Program of Jiangsu (BK20250941), the Natural Science Foundation of Nantong (grant number: JC2024026), Postgraduate Research & Practice Innovation Program of Jiangsu Province (SJCX25_2086) and Nantong University Student Innovation Training Program Project (202510304065, S202510304175).
Conflict of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Graphical abstract
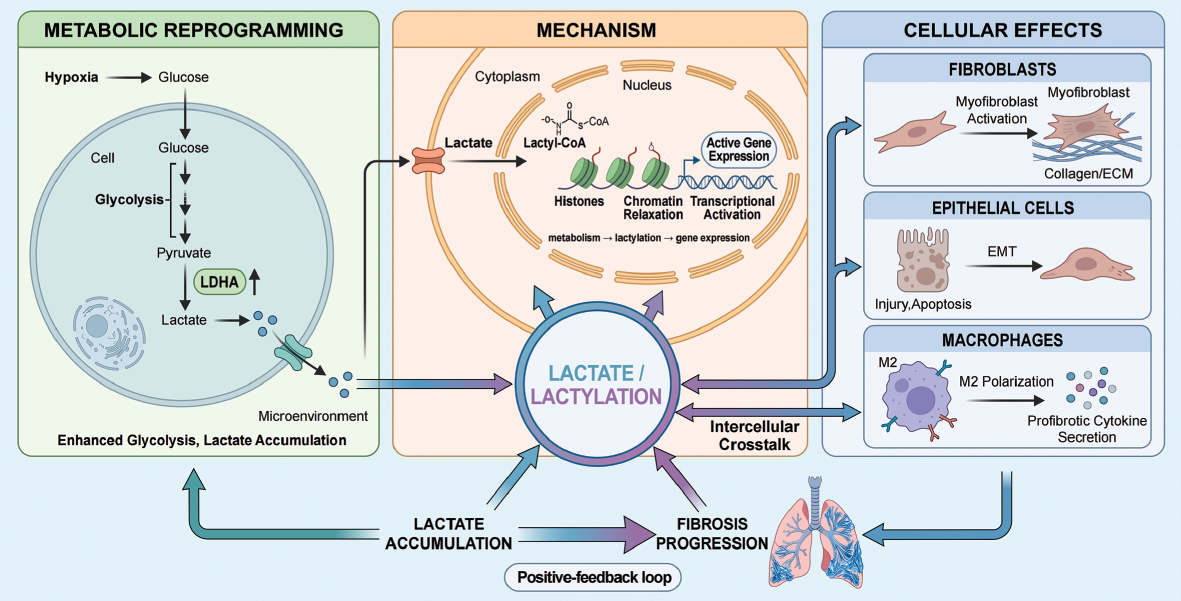
Highlights
- Lactate is redefined from a metabolic byproduct to a signaling and epigenetic regulator, with lactylation bridging metabolic reprogramming and fibrotic gene expression in pulmonary fibrosis.
- Lactylation drives key pathogenic processes, including epithelial-mesenchymal transition, macrophage polarization, fibroblast activation, and excessive extracellular matrix deposition.
- Crosstalk between lactylation and other modifications amplifies and stabilizes profibrotic signaling networks.
- The lactate-lactylation axis forms a dynamic intercellular network and represents a promising but complex therapeutic target.
In brief
Lactylation links glycolytic reprogramming to pulmonary fibrosis, redefining lactate as both a metabolic and signaling molecule. Lactate accumulation drives epigenetic and transcriptional changes that promote epithelial–mesenchymal transition, macrophage polarization, fibroblast activation, and extracellular matrix deposition. Crosstalk with other regulatory mechanisms amplifies profibrotic signaling. Targeting the lactate–lactylation axis offers therapeutic potential, though precise, cell-specific, and stage-dependent modulation remains challenging.
References
- Richeldi L, Collard HR, Jones MG. Idiopathic pulmonary fibrosis. Lancet 2017;389(10082):1941-52. [PMID: 28365056 DOI: 10.1016/s0140-6736(17)30866-8]
- Lederer DJ, Martinez FJ. Idiopathic pulmonary fibrosis. N Engl J Med 2018;378(19):1811-23. [PMID: 29742380 DOI: 10.1056/NEJMra1705751]
- Selman M, Pardo A. Idiopathic pulmonary fibrosis: an epithelial/fibroblastic cross-talk disorder. Respir Res 2001;3(1):3. [PMID: 11806838 DOI: 10.1186/rr175]
- King TE Jr, Pardo A, Selman M. Idiopathic pulmonary fibrosis. Lancet 2011;378(9807):1949-61. [PMID: 21719092 DOI: 10.1016/s0140-6736(11)60052-4]
- Cheng D, Zheng D, Jiang M, Jin Y, Liu R, et al. Inhibition of iron ion accumulation alleviates polystyrene nanoplastics-induced pulmonary fibroblast proliferation and activation. Int Immunopharmacol 2025;164:115367. [PMID: 40811949 DOI: 10.1016/j.intimp.2025.115367]
- Kim KK, Kugler MC, Wolters PJ, Robillard L, Galvez MG, et al. Alveolar epithelial cell mesenchymal transition develops in vivo during pulmonary fibrosis and is regulated by the extracellular matrix. Proc Natl Acad Sci U S A 2006;103(35):13180-5. [PMID: 16924102 DOI: 10.1073/pnas.0605669103]
- Hinz B, Phan SH, Thannickal VJ, Galli A, Bochaton-Piallat ML, et al. The myofibroblast: one function, multiple origins. Am J Pathol 2007;170(6):1807-16. [PMID: 17525249 DOI: 10.2353/ajpath.2007.070112]
- Long H, Lichtnekert J, Andrassy J, Schraml BU, Romagnani P, et al. Macrophages and fibrosis: how resident and infiltrating mononuclear phagocytes account for organ injury, regeneration or atrophy. Front Immunol 2023;14:1194988. [PMID: 37868987 DOI: 10.3389/fimmu.2023.1194988]
- JanWillem D, Lin C, Moog S, Jaillet M, Castier Y, et al. CCAAT/enhancer binding protein delta (C/EBPδ) deficiency does not affect bleomycin-induced pulmonary fibrosis. J Clin Transl Res 2018;3(Suppl 2):358-65. [PMID: 30873483 DOI: 10.18053/JCTRES.03.2017S2.004]
- Xie N, Tan Z, Banerjee S, Cui H, Ge J, et al. Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am J Respir Crit Care Med 2015;192(12):1462-74. [PMID: 26284610 DOI: 10.1164/rccm.201504-0780OC]
- Kottmann RM, Kulkarni AA, Smolnycki KA, Lyda E, Dahanayake T, et al. Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am J Respir Crit Care Med 2012;186(8):740-51. [PMID: 22923663 DOI: 10.1164/rccm.201201-0084OC]
- Yan P, Liu J, Li Z, Wang J, Zhu Z, et al. Glycolysis reprogramming in idiopathic pulmonary fibrosis: unveiling the mystery of lactate in the lung. Int J Mol Sci 2023;25(1):315. [PMID: 38203486 DOI: 10.3390/ijms25010315]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, et al. Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 2014;513(7519):559-63. [PMID: 25043024 DOI: 10.1038/nature13490]
- Fischer K, Hoffmann P, Voelkl S, Meidenbauer N, Ammer J, et al. Inhibitory effect of tumor cell–derived lactic acid on human T cells. Blood 2007;109(9):3812-9. [PMID: 17255361 DOI: 10.1182/blood-2006-07-035972]
- Hong H, Su J, Zhang Y, Xu G, Huang C, et al. A novel role of lactate: promotion of Akt-dependent elongation of microglial process. Int Immunopharmacol 2023;119:110136. [PMID: 37075668 DOI: 10.1016/j.intimp.2023.110136]
- Zhao Y, Jiang J, Zhou P, Deng K, Liu Z, et al. H3K18 lactylation-mediated VCAM1 expression promotes gastric cancer progression and metastasis via AKT-mTOR-CXCL1 axis. Biochem Pharmacol 2024;222:116120. [PMID: 38461905 DOI: 10.1016/j.bcp.2024.116120]
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, et al. Metabolic regulation of gene expression by histone lactylation. Nature 2019;574(7779):575-80. [PMID: 31645732 DOI: 10.1038/s41586-019-1678-1]
- Wan L, Zhang H, Liu J, He Q, Zhao J, et al. Lactylation and human disease. Expert Rev Mol Med 2025;27:e10. [PMID: 39895568 DOI: 10.1017/erm.2025.3]
- Xiang T, Wang X, Huang S, Zhou K, Fei S, et al. Inhibition of PKM2 by shikonin impedes TGF-β1 expression by repressing histone lactylation to alleviate renal fibrosis. Phytomedicine 2025;136:156324. [PMID: 39700636 DOI: 10.1016/j.phymed.2024.156324]
- Zong Z, Xie F, Wang S, Wu X, Zhang Z, et al. Alanyl-tRNA synthetase, AARS1, is a lactate sensor and lactyltransferase that lactylates p53 and contributes to tumorigenesis. Cell 2024;187(10):2375-92.e33. [PMID: 38653238 DOI: 10.1016/j.cell.2024.04.002]
- Nickel GA, Pederson NJ, Faheem, Yang Z, Bulf J, et al. Sirtuin 6 is a histone delactylase. J Biol Chem 2025;301(12):110795. [PMID: 41062064 DOI: 10.1016/j.jbc.2025.110795]
- Yang Y, Luo N, Gong Z, Zhou W, Ku Y, et al. Lactate and lysine lactylation of histone regulate transcription in cancer. Heliyon 2024;10(21):e38426. [PMID: 39559217 DOI: 10.1016/j.heliyon.2024.e38426]
- Merkuri F, Rothstein M, Simoes-Costa M. Histone lactylation couples cellular metabolism with developmental gene regulatory networks. Nat Commun 2024;15(1):90. [PMID: 38167340 DOI: 10.1038/s41467-023-44121-1]
- Li J, Zeng G, Zhang Z, Wang Y, Shao M, et al. Urban airborne PM2.5 induces pulmonary fibrosis through triggering glycolysis and subsequent modification of histone lactylation in macrophages. Ecotoxicol Environ Saf 2024;273:116162. [PMID: 38458067 DOI: 10.1016/j.ecoenv.2024.116162]
- Apostolova P, Pearce EL. Lactic acid and lactate: revisiting the physiological roles in the tumor microenvironment. Trends Immunol 2022;43(12):969-77. [PMID: 36319537 DOI: 10.1016/j.it.2022.10.005]
- Li X, Yang Y, Zhang B, Lin X, Fu X, et al. Lactate metabolism in human health and disease. Signal Transduct Target Ther 2022;7(1):305. [PMID: 36050306 DOI: 10.1038/s41392-022-01151-3]
- Wu D, Wang S, Wang F, Zhang Q, Zhang Z, et al. Lactate dehydrogenase A (LDHA)-mediated lactate generation promotes pulmonary vascular remodeling in pulmonary hypertension. J Transl Med 2024;22(1):738. [PMID: 39103838 DOI: 10.1186/s12967-024-05543-7]
- Vander Heiden MG, Cantley LC, Thompson CB. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 2009;324(5930):1029-33. [PMID: 19460998 DOI: 10.1126/science.1160809]
- Liberti MV, Locasale JW. The Warburg effect: how does it benefit cancer cells? Trends Biochem Sci 2016;41(3):211-8. [PMID: 26778478 DOI: 10.1016/j.tibs.2015.12.001]
- Fang C, Peng Q, Li X, Qu X, Qiao Z, et al. Multichannel immune nanoregulators suppress lactic acid metabolism and lactic acid-shaped acidic microenvironment to uproot anti-tumor immunosuppression. Adv Mater 2026;38(3):e12230. [PMID: 41001978 DOI: 10.1002/adma.202512230]
- Liu Q, Chen X, Tan Y, Liu J, Zhu M, et al. Natural products as glycolytic inhibitors for cervical cancer treatment: a comprehensive review. Biomed Pharmacother 2024;175:116708. [PMID: 38723515 DOI: 10.1016/j.biopha.2024.116708]
- Nepstad I, Hatfield KJ, Grønningsæter IS, Reikvam H. The PI3K-Akt-mTOR signaling pathway in human acute myeloid leukemia (AML) cells. Int J Mol Sci 2020;21(8):2907. [PMID: 32326335 DOI: 10.3390/ijms21082907]
- Fontana F, Giannitti G, Marchesi S, Limonta P. The PI3K/Akt pathway and glucose metabolism: a dangerous liaison in cancer. Int J Biol Sci 2024;20(8):3113-25. [PMID: 38904014 DOI: 10.7150/ijbs.89942]
- Ma Z, Ding Y, Ding X, Mou H, Mo R, et al. PDK4 rescues high-glucose-induced senescent fibroblasts and promotes diabetic wound healing through enhancing glycolysis and regulating YAP and JNK pathway. Cell Death Discov 2023;9(1):424. [PMID: 38001078 DOI: 10.1038/s41420-023-01725-2]
- Brooks GA. The science and translation of lactate shuttle theory. Cell Metab 2018;27(4):757-85. [PMID: 29617642 DOI: 10.1016/j.cmet.2018.03.008]
- Gladden LB. Lactate metabolism: a new paradigm for the third millennium. J Physiol 2004;558(Pt 1):5-30. [PMID: 15131240 DOI: 10.1113/jphysiol.2003.058701]
- Soeters PB, Shenkin A, Sobotka L, Soeters MR, de Leeuw PW, et al. The anabolic role of the Warburg, Cori-cycle and Crabtree effects in health and disease. Clin Nutr 2021;40(5):2988-98. [PMID: 33674148 DOI: 10.1016/j.clnu.2021.02.012]
- Mao Y, Zhang J, Zhou Q, He X, Zheng Z, et al. Hypoxia induces mitochondrial protein lactylation to limit oxidative phosphorylation. Cell Res 2024;34(1):13-30. [PMID: 38163844 DOI: 10.1038/s41422-023-00864-6]
- Gaffney DO, Jennings EQ, Anderson CC, Marentette JO, Shi T, et al. Non-enzymatic lysine lactoylation of glycolytic enzymes. Cell Chem Biol 2020;27(2):206-13.e6. [PMID: 31767537 DOI: 10.1016/j.chembiol.2019.11.005]
- Gonzatti MB, Hintzen JCJ, Sharma I, Najar MA, Tsusaka T, et al. Class I histone deacetylases catalyze lysine lactylation. J Biol Chem 2025;301(10):110602. [PMID: 40835008 DOI: 10.1016/j.jbc.2025.110602]
- Sun Y, Wang H, Cui Z, Yu T, Song Y, et al. Lactylation in cancer progression and drug resistance. Drug Resist Updat 2025;81:101248. [PMID: 40287994 DOI: 10.1016/j.drup.2025.101248]
- Brooks GA. The tortuous path of lactate shuttle discovery: from cinders and boards to the lab and ICU. J Sport Health Sci 2020;9(5):446-60. [PMID: 32444344 DOI: 10.1016/j.jshs.2020.02.006]
- Wang X, Liu H, Ni Y, Shen P, Han X. Lactate shuttle: from substance exchange to regulatory mechanism. Hum Cell 2022;35(1):1-14. [PMID: 34606041 DOI: 10.1007/s13577-021-00622-z]
- Brooks GA, Curl CC, Leija RG, Osmond AD, Duong JJ, et al. Tracing the lactate shuttle to the mitochondrial reticulum. Exp Mol Med 2022;54(9):1332-47. [PMID: 36075947 DOI: 10.1038/s12276-022-00802-3]
- Natsubori A, Hirai S, Kwon S, Ono D, Deng F, et al. Serotonergic neurons control cortical neuronal intracellular energy dynamics by modulating astrocyte-neuron lactate shuttle. iScience 2023;26(1):105830. [PMID: 36713262 DOI: 10.1016/j.isci.2022.105830]
- Wei T, Guo Y, Huang C, Sun M, Zhou B, et al. Fibroblast-to-cardiomyocyte lactate shuttle modulates hypertensive cardiac remodelling. Cell Biosci 2023;13(1):151. [PMID: 37580825 DOI: 10.1186/s13578-023-01098-0]
- Chen L, Lin Y, Zhu X, Zhuo S, Li Z, et al. MCT1-mediated lactate shuttle to mitochondria governs macrophage polarization and modulates glucose homeostasis by affecting β cells. Adv Sci (Weinh) 2025;12(38):e14760. [PMID: 40660708 DOI: 10.1002/advs.202414760]
- D’Aria S, Maquet C, Li S, Dhup S, Lepez A, et al. Expression of the monocarboxylate transporter MCT1 is required for virus-specific mouse CD8+ T cell memory development. Proc Natl Acad Sci U S A 2024;121(13):e2306763121. [PMID: 38498711 DOI: 10.1073/pnas.2306763121]
- Menig-Benzig LS, Stühler V, Mazzola P, Heinrich H, Hofmann U, et al. Drug targeting of the monocarboxylate transporter MCT4 is a novel treatment strategy for metastatic ccRCC. Pharmacol Res 2025;222:108057. [PMID: 41349849 DOI: 10.1016/j.phrs.2025.108057]
- Fang Y, Liu W, Tang Z, Ji X, Zhou Y, et al. Monocarboxylate transporter 4 inhibition potentiates hepatocellular carcinoma immunotherapy through enhancing T cell infiltration and immune attack. Hepatology 2023;77(1):109-23. [PMID: 35043976 DOI: 10.1002/hep.32348]
- Ma M, Liu T, Chen H, Wang Z, Zhou J, et al. Monocarboxylate transporter 4 inhibition reduces synovial hyperproliferation and metabolic reprogramming under hypoxia in rheumatoid arthritis. Arch Med Res 2025;57(1):103283. [PMID: 40829205 DOI: 10.1016/j.arcmed.2025.103283]
- Jha MK, Passero JV, Rawat A, Ament XH, Yang F, et al. Macrophage monocarboxylate transporter 1 promotes peripheral nerve regeneration after injury in mice. J Clin Invest 2021;131(21):e141964. [PMID: 34491913 DOI: 10.1172/JCI141964]
- Ahmed K, Tunaru S, Tang C, Müller M, Gille A, et al. An autocrine lactate loop mediates insulin-dependent inhibition of lipolysis through GPR81. Cell Metab 2010;11(4):311-9. [PMID: 20374963 DOI: 10.1016/j.cmet.2010.02.012]
- Pellerin L, Magistretti PJ. Glutamate uptake into astrocytes stimulates aerobic glycolysis: a mechanism coupling neuronal activity to glucose utilization. Proc Natl Acad Sci U S A 1994;91(22):10625-9. [PMID: 7938003 DOI: 10.1073/pnas.91.22.10625]
- Liu C, Wu J, Zhu J, Kuei C, Yu J, et al. Lactate inhibits lipolysis in fat cells through activation of an orphan G-protein-coupled receptor, GPR81. J Biol Chem 2009;284(5):2811-22. [PMID: 19047060 DOI: 10.1074/jbc.M806409200]
- Wynn TA. Cellular and molecular mechanisms of fibrosis. J Pathol 2008;214(2):199-210. [PMID: 18161745 DOI: 10.1002/path.2277]
- Hinz B. Formation and function of the myofibroblast during tissue repair. J Invest Dermatol 2007;127(3):526-37. [PMID: 17299435 DOI: 10.1038/sj.jid.5700613]
- Wang LJ, Guo PF, Chen YZ, Yan HW, Zhang XL. Tangeretin in the treatment of pulmonary fibrosis: current advances and future perspectives. Curr Mol Pharmacol 2024;17:e18761429385520. [PMID: 40370245 DOI: 10.2174/0118761429385520250508041438]
- Distler JHW, Györfi AH, Ramanujam M, Whitfield ML, Königshoff M, et al. Shared and distinct mechanisms of fibrosis. Nat Rev Rheumatol 2019;15(12):705-30. [PMID: 31712723 DOI: 10.1038/s41584-019-0322-7]
- Peng D, Fu M, Wang M, Wei Y, Wei X. Targeting TGF-β signal transduction for fibrosis and cancer therapy. Mol Cancer 2022;21(1):104. [PMID: 35461253 DOI: 10.1186/s12943-022-01569-x]
- Bernard K, Logsdon NJ, Ravi S, Xie N, Persons BP, et al. Metabolic reprogramming is required for myofibroblast contractility and differentiation. J Biol Chem 2015;290(42):25427-38. [PMID: 26318453 DOI: 10.1074/jbc.M115.646984]
- Higgins DF, Kimura K, Bernhardt WM, Shrimanker N, Akai Y, et al. Hypoxia promotes fibrogenesis in vivo via HIF-1 stimulation of epithelial-to-mesenchymal transition. J Clin Invest 2007;117(12):3810-20. [PMID: 18037992 DOI: 10.1172/JCI30487]
- Noble PW, Albera C, Bradford WZ, Costabel U, Glassberg MK, et al. Pirfenidone in patients with idiopathic pulmonary fibrosis (CAPACITY): two randomised trials. Lancet 2011;377(9779):1760-9. [PMID: 21571362 DOI: 10.1016/S0140-6736(11)60405-4]
- Richeldi L, du Bois RM, Raghu G, Azuma A, Brown KK, et al. Efficacy and safety of nintedanib in idiopathic pulmonary fibrosis. N Engl J Med 2014;370(22):2071-82. [PMID: 24836310 DOI: 10.1056/NEJMoa1402584]
- Shen Y, Li X, Wei D, Guo H, Yang X, et al. Synergistic effects of diammonium glycyrrhizinate and vitamin D3 against EMT in pulmonary fibrosis through modulating the crosstalk of STAT3/HSP90AA1 and HIF-1α pathway. J Ethnopharmacol 2026;359:120649. [PMID: 41423159 DOI: 10.1016/j.jep.2025.120649]
- Zhang X, Zhang X, Huang W, Ge X. The role of heat shock proteins in the regulation of fibrotic diseases. Biomed Pharmacother 2021;135:111067. [PMID: 33383375 DOI: 10.1016/j.biopha.2020.111067]
- Newton DA, Lottes RG, Ryan RM, Spyropoulos DD, Baatz JE. Dysfunctional lactate metabolism in human alveolar type II cells from idiopathic pulmonary fibrosis lung explant tissue. Respir Res 2021;22(1):278. [PMID: 34711218 DOI: 10.1186/s12931-021-01866-x]
- Parimon T, Yao C, Stripp BR, Noble PW, Chen P. Alveolar epithelial type II cells as drivers of lung fibrosis in idiopathic pulmonary fibrosis. Int J Mol Sci 2020;21(7):2269. [PMID: 32218238 DOI: 10.3390/ijms21072269]
- Feng J, Zhong H, Mei S, Tang R, Zhou Y, et al. LPS-induced monocarboxylate transporter-1 inhibition facilitates lactate accumulation triggering epithelial-mesenchymal transformation and pulmonary fibrosis. Cell Mol Life Sci 2024;81(1):206. [PMID: 38709307 DOI: 10.1007/s00018-024-05242-y]
- Sun Z, He W, Meng H, Ji Z, Qu J, et al. Lactate activates ER stress to promote alveolar epithelial cells apoptosis in pulmonary fibrosis. Respir Res 2024;25(1):401. [PMID: 39522031 DOI: 10.1186/s12931-024-03016-5]
- Wang P, Xie D, Xiao T, Cheng C, Wang D, et al. H3K18 lactylation promotes the progression of arsenite-related idiopathic pulmonary fibrosis via YTHDF1/m6A/NREP. J Hazard Mater 2024;461:132582. [PMID: 37742376 DOI: 10.1016/j.jhazmat.2023.132582]
- Liu S, He Y, Jin L, Shi S, Zhang J, et al. H3K18 lactylation-mediated SIX1 upregulation contributes to silica-induced epithelial-mesenchymal transition in airway epithelial cells. Toxicology 2025;514:154109. [PMID: 40049282 DOI: 10.1016/j.tox.2025.154109]
- Wei Y, Guo H, Chen S, Tang XX. Regulation of macrophage activation by lactylation in lung disease. Front Immunol 2024;15:1427739. [PMID: 39026681 DOI: 10.3389/fimmu.2024.1427739]
- Yan P, Yang K, Xu M, Zhu M, Duan Y, et al. CCT6A alleviates pulmonary fibrosis by inhibiting HIF-1α-mediated lactate production. J Mol Cell Biol 2024;16(5):mjae021. [PMID: 38760881 DOI: 10.1093/jmcb/mjae021]
- Wang L, Yuan H, Li W, Yan P, Zhao M, et al. ACSS3 regulates the metabolic homeostasis of epithelial cells and alleviates pulmonary fibrosis. Biochim Biophys Acta Mol Basis Dis 2024;1870(2):166960. [PMID: 37979225 DOI: 10.1016/j.bbadis.2023.166960]
- Callaghan NI, Davenport Huyer L. Altered metabolism in idiopathic pulmonary fibrosis. J Cell Physiol 2025;240(12):e70112. [PMID: 41424408 DOI: 10.1002/jcp.70112]
- Gao H, Sun Z, Hu X, Song W, Liu Y, et al. Identification of glycolysis-related gene signatures for prognosis and therapeutic targeting in idiopathic pulmonary fibrosis. Front Pharmacol 2025;16:1486357. [PMID: 40093327 DOI: 10.3389/fphar.2025.1486357]
- Gui X, Qiu X, Tian Y, Xie M, Li H, et al. Prognostic value of IFN-γ, sCD163, CCL2 and CXCL10 involved in acute exacerbation of idiopathic pulmonary fibrosis. Int Immunopharmacol 2019;70:208-15. [PMID: 30851700 DOI: 10.1016/j.intimp.2019.02.039]
- Liu Z, Liu W, Wei H, Ping Y, Yu Z, et al. Elevated lactate production exacerbates PM2.5-induced pulmonary fibrosis by stabilizing TGF-β1. J Adv Res 2026;83:299-314. [PMID: 40759348 DOI: 10.1016/j.jare.2025.07.057]
- Xie N, Cui H, Ge J, Banerjee S, Guo S, et al. Metabolic characterization and RNA profiling reveal glycolytic dependence of profibrotic phenotype of alveolar macrophages in lung fibrosis. Am J Physiol Lung Cell Mol Physiol 2017;313(5):L834-44. [PMID: 28798256 DOI: 10.1152/ajplung.00235.2017]
- Xu Q, Mei S, Nie F, Zhang Z, Feng J, et al. The role of macrophage–fibroblast interaction in lipopolysaccharide-induced pulmonary fibrosis: an acceleration in lung fibroblast aerobic glycolysis. Lab Invest 2022;102(4):432-9. [PMID: 34775492 DOI: 10.1038/s41374-021-00701-7]
- Cui H, Xie N, Banerjee S, Ge J, Jiang D, et al. Lung myofibroblasts promote macrophage profibrotic activity through lactate-induced histone lactylation. Am J Respir Cell Mol Biol 2021;64(1):115-25. [PMID: 33074715 DOI: 10.1165/rcmb.2020-0360OC]
- Shi L, Zhang L, Ma Z, Hu Z, Geng Z, et al. H3K18 lactylation drives the progression of silica nanoparticles-induced pulmonary fibrosis via promoting macrophage M1 polarization. J Hazard Mater 2025;496:139286. [PMID: 40690843 DOI: 10.1016/j.jhazmat.2025.139286]
- Wang H, Gao Y, Wang L, Yu Y, Zhang J, et al. Lung specific homing of diphenyleneiodonium chloride improves pulmonary fibrosis by inhibiting macrophage M2 metabolic program. J Adv Res 2023;44:213-25. [PMID: 36725191 DOI: 10.1016/j.jare.2022.04.012]
- Shinoda H, Tasaka S, Fujishima S, Yamasawa W, Miyamoto K, et al. Elevated CC chemokine level in bronchoalveolar lavage fluid is predictive of a poor outcome of idiopathic pulmonary fibrosis. Respiration 2009;78(3):285-92. [PMID: 19270434 DOI: 10.1159/000207617]
- Hu Y, Yang L, Huang L, Zeng C, Ren S. m6A reader IGF2BP1 facilitates macrophage glycolytic metabolism and fibrotic phenotype by stabilizing THBS1 mRNA to promote pulmonary fibrosis. Cell Mol Life Sci 2025;82(1):157. [PMID: 40220148 DOI: 10.1007/s00018-025-05673-1]
- Liu C, Zhang Q, Zhou H, Jin L, Liu C, et al. GLP-1R activation attenuates the progression of pulmonary fibrosis via disrupting NLRP3 inflammasome/PFKFB3-driven glycolysis interaction and histone lactylation. J Transl Med 2024;22(1):954. [PMID: 39434134 DOI: 10.1186/s12967-024-05753-z]
- Zhao G, Zhou J, He S, Fei X, Guo G. The role of lactylation in virus–host interactions. Int J Mol Sci 2025;26(14):6613. [PMID: 40724863 DOI: 10.3390/ijms26146613]
- Rho H, Hay N. Protein lactylation in cancer: mechanisms and potential therapeutic implications. Exp Mol Med 2025;57(3):545-53. [PMID: 40128358 DOI: 10.1038/s12276-025-01410-7]
- Shi P, Ma Y, Zhang S. Non-histone lactylation: unveiling its functional significance. Front Cell Dev Biol 2025;13:1535611. [PMID: 39925738 DOI: 10.3389/fcell.2025.1535611]
- Deng YT, Ma L, Mei Y, Wang JS, Bai XH, et al. Amphiregulin contributes to neuropathic pain by enhancing glycolysis that stimulates histone lactylation in sensory neurons. Sci Signal 2025;18(891):eadr9397. [PMID: 40526786 DOI: 10.1126/scisignal.adr9397]
- Liu J, Zhao L, Yan M, Jin S, Shang L, et al. H4K79 and H4K91 histone lactylation, newly identified lactylation sites enriched in breast cancer. J Exp Clin Cancer Res 2025;44(1):252. [PMID: 40849487 DOI: 10.1186/s13046-025-03512-6]
- Zhu R, Ye X, Lu X, Xiao L, Yuan M, et al. ACSS2 acts as a lactyl-CoA synthetase and couples KAT2A to function as a lactyltransferase for histone lactylation and tumor immune evasion. Cell Metab 2025;37(2):361-76.e7. [PMID: 39561764 DOI: 10.1016/j.cmet.2024.10.015]
- Chen J, He J, Wang X, Bai L, Yang X, et al. Glis1 inhibits RTEC cellular senescence and renal fibrosis by downregulating histone lactylation in DKD. Life Sci 2025;361:123293. [PMID: 39643036 DOI: 10.1016/j.lfs.2024.123293]
- Xie B, Zhang M, Li J, Cui J, Zhang P, et al. KAT8-catalyzed lactylation promotes eEF1A2-mediated protein synthesis and colorectal carcinogenesis. Proc Natl Acad Sci U S A 2024;121(8):e2314128121. [PMID: 38359291 DOI: 10.1073/pnas.2314128121]
- Niu Z, Chen C, Wang S, Lu C, Wu Z, et al. HBO1 catalyzes lysine lactylation and mediates histone H3K9la to regulate gene transcription. Nat Commun 2024;15(1):3561. [PMID: 38670996 DOI: 10.1038/s41467-024-47900-6]
- Yin D, Jiang N, Cheng C, Sang X, Feng Y, et al. Protein lactylation and metabolic regulation of the zoonotic parasite Toxoplasma gondii. Genomics Proteomics Bioinformatics 2023;21(6):1163-81. [PMID: 36216028 DOI: 10.1016/j.gpb.2022.09.010]
- Sun S, Xu Z, He L, Shen Y, Yan Y, et al. Metabolic regulation of cytoskeleton functions by HDAC6-catalyzed α-tubulin lactylation. Nat Commun 2024;15(1):8377. [PMID: 39333081 DOI: 10.1038/s41467-024-52729-0]
- Wang J, Wang Z, Wang Q, Li X, Guo Y. Ubiquitous protein lactylation in health and diseases. Cell Mol Biol Lett 2024;29(1):23. [PMID: 38317138 DOI: 10.1186/s11658-024-00541-5]
- Wang Z, Liu Z, Lv M, Luan Z, Li T, et al. Novel histone modifications and liver cancer: emerging frontiers in epigenetic regulation. Clin Epigenetics 2025;17(1):30. [PMID: 39980025 DOI: 10.1186/s13148-025-01838-8]
- Ma C, Liao R, Chen X, Cao Q, Deng X, et al. RHOA lactylation at oncogenic hotspots promotes oncogenic activity and protein stabilization. Mol Cancer 2025;24(1):311. [PMID: 41291745 DOI: 10.1186/s12943-025-02511-7]
- Zhang TN, Huang XM, Li L, Li Y, Liu YP, et al. Lactylation of HADHA promotes sepsis-induced myocardial depression. Circ Res 2025;137(4):e65-87. [PMID: 40575877 DOI: 10.1161/CIRCRESAHA.124.325708]
- Zhang X, Liu Y, Rekowski MJ, Wang N. Lactylation of tau in human Alzheimer’s disease brains. Alzheimers Dement 2025;21(2):e14481. [PMID: 39740133 DOI: 10.1002/alz.14481]
- Wang Y, Li H, Jiang S, Fu D, Lu X, et al. The glycolytic enzyme PFKFB3 drives kidney fibrosis through promoting histone lactylation-mediated NF-κB family activation. Kidney Int 2024;106(2):226-40. [PMID: 38789037 DOI: 10.1016/j.kint.2024.04.016]
- Zhang C, Zhou T, Li C, Wang D, Tao J, et al. Deciphering novel enzymatic and non-enzymatic lysine lactylation in Salmonella. Emerg Microbes Infect 2025;14(1):2475838. [PMID: 40035788 DOI: 10.1080/22221751.2025.2475838]
- Trujillo MN, Jennings EQ, Hoffman EA, Zhang H, Phoebe AM, et al. Lactoylglutathione promotes inflammatory signaling in macrophages through histone lactoylation. Mol Metab 2024;81:101888. [PMID: 38307385 DOI: 10.1016/j.molmet.2024.101888]
- Clément C, Dorion S, Bykova NV, Fetterley V, Branchini E, et al. Implication of S-d-lactoylglutathione in the spontaneous cysteine S-glutathionylation and lysine N-lactoylation of Arabidopsis thaliana NAD-dependent glyceraldehyde-3-phosphate dehydrogenase. Int J Mol Sci 2025;26(19):9673. [PMID: 41096938 DOI: 10.3390/ijms26199673]
- Moreno-Yruela C, Bæk M, Monda F, Olsen CA. Chiral posttranslational modification to lysine ε-amino groups. Acc Chem Res 2022;55(10):1456-66. [PMID: 35500056 DOI: 10.1021/acs.accounts.2c00115]
- Li F, Si W, Xia L, Yin D, Wei T, et al. Positive feedback regulation between glycolysis and histone lactylation drives oncogenesis in pancreatic ductal adenocarcinoma. Mol Cancer 2024;23(1):90. [PMID: 38711083 DOI: 10.1186/s12943-024-02008-9]
- Lv J, Yu X, Liu X, Zhang Q, Zhang M, et al. The LncRNA STEAP3-AS1 promotes liver metastasis in colorectal cancer by regulating histone lactylation through chromatin remodelling. J Exp Clin Cancer Res 2025;44(1):205. [PMID: 40665344 DOI: 10.1186/s13046-025-03461-0]
- Chen H, Li Y, Li H, Chen X, Fu H, et al. NBS1 lactylation is required for efficient DNA repair and chemotherapy resistance. Nature 2024;631(8021):663-9. [PMID: 38961290 DOI: 10.1038/s41586-024-07620-9]
- Lin J, Yin Y, Cao J, Zhang Y, Chen J, et al. NUDT21 lactylation reprograms alternative polyadenylation to promote cuproptosis resistance. Cell Discov 2025;11(1):52. [PMID: 40425546 DOI: 10.1038/s41421-025-00804-1]
- Moreno-Yruela C, Zhang D, Wei W, Bæk M, Liu W, et al. Class I histone deacetylases (HDAC1–3) are histone lysine delactylases. Sci Adv 2022;8(3):eabi6696. [PMID: 35044827 DOI: 10.1126/sciadv.abi6696]
- Fan Z, Liu Z, Zhang N, Wei W, Cheng K, et al. Identification of SIRT3 as an eraser of H4K16la. iScience 2023;26(10):107757. [PMID: 37720100 DOI: 10.1016/j.isci.2023.107757]
- Fan W, Zeng S, Wang X, Wang G, Liao D, et al. A feedback loop driven by H3K9 lactylation and HDAC2 in endothelial cells regulates VEGF-induced angiogenesis. Genome Biol 2024;25(1):165. [PMID: 38918851 DOI: 10.1186/s13059-024-03308-5]
- Zou Y, Cao M, Tai M, Zhou H, Tao L, et al. A feedback loop driven by H4K12 lactylation and HDAC3 in macrophages regulates lactate-induced collagen synthesis in fibroblasts via the TGF-β signaling. Adv Sci (Weinh) 2025;12(13):e2411408. [PMID: 39945346 DOI: 10.1002/advs.202411408]
- Zhou J, Qiu S, Yang X, Wu Y, Yao X, et al. Hypoxia-induced PRMT1 lactylation drives vimentin arginine asymmetric dimethylation in tumor metastasis. Adv Sci (Weinh) 2025;12(41):e09861. [PMID: 40884268 DOI: 10.1002/advs.202509861]
- Tsukihara S, Akiyama Y, Shimada S, Hatano M, Igarashi Y, et al. Delactylase effects of SIRT1 on a positive feedback loop involving the H19-glycolysis-histone lactylation in gastric cancer. Oncogene 2025;44(11):724-38. [PMID: 39658647 DOI: 10.1038/s41388-024-03243-6]
- Sun Y, Chen Y, Xu Y, Zhang Y, Lu M, et al. Genetic encoding of ε–N–l-lactyllysine for detecting delactylase activity in living cells. Chem Commun (Camb) 2022;58(61):8544-7. [PMID: 35815577 DOI: 10.1039/d2cc02643k]
- Zhang N, Zhang Y, Xu J, Wang P, Wu B, et al. α-myosin heavy chain lactylation maintains sarcomeric structure and function and alleviates the development of heart failure. Cell Res 2023;33(9):679-98. [PMID: 37443257 DOI: 10.1038/s41422-023-00844-w]
- Chen C, Zhang Y, Zang Y, Fan Z, Han Y, et al. SIRT3 functions as an eraser of histone H3K9 lactylation to modulate transcription for inhibiting the progression of esophageal cancer. Mol Cell Proteomics 2025;24(5):100973. [PMID: 40252727 DOI: 10.1016/j.mcpro.2025.100973]
- Wang ZA, Markert J, Whedon SD, Abeywardana MY, Sheng X, et al. Structural and enzymatic plasticity of SIRT6 deacylase activity. J Biol Chem 2025;301(5):108446. [PMID: 40147774 DOI: 10.1016/j.jbc.2025.108446]
- Hu X, Huang X, Yang Y, Sun Y, Zhao Y, et al. Dux activates metabolism-lactylation-MET network during early iPSC reprogramming with Brg1 as the histone lactylation reader. Nucleic Acids Res 2024;52(10):5529-48. [PMID: 38512058 DOI: 10.1093/nar/gkae183]
- Zhai G, Niu Z, Jiang Z, Zhao F, Wang S, et al. DPF2 reads histone lactylation to drive transcription and tumorigenesis. Proc Natl Acad Sci U S A 2024;121(50):e2421496121. [PMID: 39636855 DOI: 10.1073/pnas.2421496121]
- Nuñez R, Sidlowski PFW, Steen EA, Wynia-Smith SL, Sprague DJ, et al. The TRIM33 bromodomain recognizes histone lysine lactylation. ACS Chem Biol 2024;19(12):2418-28. [PMID: 39556662 DOI: 10.1021/acschembio.4c00248]
- Dai C, Tang Y, Yang H, Zheng J. YTHDC1 lactylation regulates its phase separation to enhance target mRNA stability and promote RCC progression. Mol Cell 2025;85(14):2733-48.e7. [PMID: 40680722 DOI: 10.1016/j.molcel.2025.06.017]
- Iozzo M, Pardella E, Giannoni E, Chiarugi P. The role of protein lactylation: a kaleidoscopic post-translational modification in cancer. Mol Cell 2025;85(7):1263-79. [PMID: 40073861 DOI: 10.1016/j.molcel.2025.02.011]
- Yu X, Yang J, Xu J, Pan H, Wang W, et al. Histone lactylation: from tumor lactate metabolism to epigenetic regulation. Int J Biol Sci 2024;20(5):1833-54. [PMID: 38481814 DOI: 10.7150/ijbs.91492]
- Liu R, Ren X, Park YE, Feng H, Sheng X, et al. Nuclear GTPSCS functions as a lactyl-CoA synthetase to promote histone lactylation and gliomagenesis. Cell Metab 2025;37(2):377-94.e9. [PMID: 39642882 DOI: 10.1016/j.cmet.2024.11.005]
- Sui Y, Shen Z, Wang Z, Feng J, Zhou G. Lactylation in cancer: metabolic mechanism and therapeutic strategies. Cell Death Discov 2025;11(1):68. [PMID: 39979245 DOI: 10.1038/s41420-025-02349-4]
- Ziogas A, Novakovic B, Ventriglia L, Galang N, Tran KA, et al. Long-term histone lactylation connects metabolic and epigenetic rewiring in innate immune memory. Cell 2025;188(11):2992-3012.e16. [PMID: 40318634 DOI: 10.1016/j.cell.2025.03.048]
- Raychaudhuri D, Singh P, Chakraborty B, Hennessey M, Tannir AJ, et al. Histone lactylation drives CD8+ T cell metabolism and function. Nat Immunol 2024;25(11):2140-51. [PMID: 39375549 DOI: 10.1038/s41590-024-01985-9]
- Yang J, Yu X, Xiao M, Xu H, Tan Z, et al. Histone lactylation-driven feedback loop modulates cholesterol-linked immunosuppression in pancreatic cancer. Gut 2025;74(11):1859-72. [PMID: 40467104 DOI: 10.1136/gutjnl-2024-334361]
- Wang S, Huang T, Wu Q, Yuan H, Wu X, et al. Lactate reprograms glioblastoma immunity through CBX3-regulated histone lactylation. J Clin Invest 2024;134(22):e176851. [PMID: 39545414 DOI: 10.1172/jci176851]
- Lv M, Yang X, Xu C, Song Q, Zhao H, et al. SIRT4 promotes pancreatic cancer stemness by enhancing histone lactylation and epigenetic reprogramming stimulated by calcium signaling. Adv Sci (Weinh) 2025;12(20):e2412553. [PMID: 40298941 DOI: 10.1002/advs.202412553]
- Huang Y, Wang C, Zhou T, Xie F, Liu Z, et al. Lumican promotes calcific aortic valve disease through H3 histone lactylation. Eur Heart J 2024;45(37):3871-85. [PMID: 38976370 DOI: 10.1093/eurheartj/ehae407]
- Zhang Y, Song H, Li M, Lu P. Histone lactylation bridges metabolic reprogramming and epigenetic rewiring in driving carcinogenesis: oncometabolite fuels oncogenic transcription. Clin Transl Med 2024;14(3):e1614. [PMID: 38456209 DOI: 10.1002/ctm2.1614]
- Wang N, Wang W, Wang X, Mang G, Chen J, et al. Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ Res 2022;131(11):893-908. [PMID: 36268709 DOI: 10.1161/CIRCRESAHA.122.320488]
- Zhang J, Wu D, Zeng F, Gu H, Li C, et al. Lactate metabolic reprogramming and histone lactylation modification in sepsis. Int J Biol Sci 2025;21(11):5034-55. [PMID: 40860191 DOI: 10.7150/ijbs.116088]
- Zhang Q, Han Z, Liu JH, Li M, Li W, et al. Hypoxia facilitates stemness of colon cancer cells via histone lactylation. Biochim Biophys Acta Mol Basis Dis 2025;1871(8):167993. [PMID: 40716503 DOI: 10.1016/j.bbadis.2025.167993]
- Zhao SS, Liu J, Wu QC, Zhou XL. Lactate regulates pathological cardiac hypertrophy via histone lactylation modification. J Cell Mol Med 2024;28(16):e70022. [PMID: 39205384 DOI: 10.1111/jcmm.70022]
- Chen A, Chen Z, Huang B, Lian G, Luo L, et al. Hypoxia-induced histone lactylation promotes pulmonary arterial smooth muscle cells proliferation in pulmonary hypertension. Mol Cell Biochem 2025;480(11):5685-97. [PMID: 40588665 DOI: 10.1007/s11010-025-05342-8]
- Zhang YM, Yang F, Li Q, Zhang JN. Role of histone lactylation in neurological disorders. Int J Mol Sci 2025;26(16):7949. [PMID: 40869269 DOI: 10.3390/ijms26167949]
- Liang Z, Liu J, Gou Y, Wang H, Li Z, et al. Elevated histone lactylation mediates ferroptosis resistance in endometriosis through the METTL3-regulated HIF1A/HMOX1 signaling pathway. Adv Sci (Weinh) 2025;12(31):e08220. [PMID: 40492577 DOI: 10.1002/advs.202408220]
- Liu Y, Guo X, Hu X, Zhou S, Yang Q, et al. Lysine lactylation in diseases: beyond histone lactylation. Cell Death Dis 2025;17(1):55. [PMID: 41285721 DOI: 10.1038/s41419-025-08223-6]
- Zhang Q, Luo B, Sun X, Nagashima H, Wu Y, et al. Non-histone lysine lactylation: emerging roles in tumor biology and therapeutic implications. Ageing Res Rev 2025;112:102875. [PMID: 40825375 DOI: 10.1016/j.arr.2025.102875]
- Kim JY, Sim H, Na AY, Choi SY, Lee S. Global profiling of lysine lactylation in prostate cancer cells. Cancer Genomics Proteomics 2025;22(6):929-39. [PMID: 41151862 DOI: 10.21873/cgp.20547]
- Yang YH, Wang QC, Kong J, Yang JT, Liu JF. Global profiling of lysine lactylation in human lungs. Proteomics 2023;23(15):e2200437. [PMID: 37170646 DOI: 10.1002/pmic.202200437]
- Dong H, Zhang J, Zhang H, Han Y, Lu C, et al. YiaC and CobB regulate lysine lactylation in Escherichia Coli. Nat Commun 2022;13(1):6628. [PMID: 36333310 DOI: 10.1038/s41467-022-34399-y]
- Peng X, Du J. Histone and non-histone lactylation: molecular mechanisms, biological functions, diseases, and therapeutic targets. Mol Biomed 2025;6(1):38. [PMID: 40484921 DOI: 10.1186/s43556-025-00275-6]
- Li S, Dong L, Wang K. Current and future perspectives of lysine lactylation in cancer. Trends Cell Biol 2025;35(3):190-3. [PMID: 39837737 DOI: 10.1016/j.tcb.2024.12.015]
- Yang Z, Zheng Y, Gao Q. Lysine lactylation in the regulation of tumor biology. Trends Endocrinol Metab 2024;35(8):720-31. [PMID: 38395657 DOI: 10.1016/j.tem.2024.01.011]
- Gao X, Pang C, Fan Z, Wang Y, Duan Y, et al. Regulation of newly identified lysine lactylation in cancer. Cancer Lett 2024;587:216680. [PMID: 38346584 DOI: 10.1016/j.canlet.2024.216680]
- Chen J, Feng Q, Qiao Y, Pan S, Liang L, et al. ACSF2 and lysine lactylation contribute to renal tubule injury in diabetes. Diabetologia 2024;67(7):1429-43. [PMID: 38676722 DOI: 10.1007/s00125-024-06156-x]
- Fan M, Yang K, Wang X, Chen L, Gill PS, et al. Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci Adv 2023;9(5):eadc9465. [PMID: 36735787 DOI: 10.1126/sciadv.adc9465]
- Pan RY, He L, Zhang J, Liu X, Liao Y, et al. Positive feedback regulation of microglial glucose metabolism by histone H4 lysine 12 lactylation in Alzheimer’s disease. Cell Metab 2022;34(4):634-48.e6. [PMID: 35303422 DOI: 10.1016/j.cmet.2022.02.013]
- Llibre A, Kucuk S, Gope A, Certo M, Mauro C. Lactate: a key regulator of the immune response. Immunity 2025;58(3):535-54. [PMID: 40073846 DOI: 10.1016/j.immuni.2025.02.008]
- Yang Y, Deng L, Zhang H, Hu J, Li J, et al. Function and mechanism of lactylation in health and diseases: a comprehensive review. Int J Surg 2025;111(8):5387-402. [PMID: 40511917 DOI: 10.1097/js9.0000000000002623]
- Susser LI, Nguyen MA, Geoffrion M, Emerton C, Ouimet M, et al. Mitochondrial fragmentation promotes inflammation resolution responses in macrophages via histone lactylation. Mol Cell Biol 2023;43(10):531-46. [PMID: 37807652 DOI: 10.1080/10985549.2023.2253131]
- You X, Xie Y, Tan Q, Zhou C, Gu P, et al. Glycolytic reprogramming governs crystalline silica-induced pyroptosis and inflammation through promoting lactylation modification. Ecotoxicol Environ Saf 2024;283:116952. [PMID: 39217895 DOI: 10.1016/j.ecoenv.2024.116952]
- Zou W, Zhang Z, Cao T, Li M. Mesenchymal stem cell transplantation ameliorates inflammation in spinal cord injury by inhibiting lactylation-related genes. Cytokine 2025;191:156960. [PMID: 40345018 DOI: 10.1016/j.cyto.2025.156960]
- Al-Malsi K, Xie S, Cai Y, Mohammed N, Xie K, et al. The role of lactylation in tumor growth and cancer progression. Front Oncol 2025;15:1516785. [PMID: 39968078 DOI: 10.3389/fonc.2025.1516785]
- Li W, Zhou J, Gu Y, Chen Y, Huang Y, et al. Lactylation of RNA m6A demethylase ALKBH5 promotes innate immune response to DNA herpesviruses and mpox virus. Proc Natl Acad Sci U S A 2024;121(43):e2409132121. [PMID: 39413129 DOI: 10.1073/pnas.2409132121]
- Wang X, Yan X, Mang G, Chen Y, Liu S, et al. S100a9 lactylation triggers neutrophil trafficking and cardiac inflammation in myocardial ischemia/reperfusion injury. J Clin Invest 2025;135(24):e194664. [PMID: 41066195 DOI: 10.1172/jci194664]
- Wu X, Liu C, Zhang C, Kuai L, Hu S, et al. The role of lactate and lactylation in the dysregulation of immune responses in psoriasis. Clin Rev Allergy Immunol 2025;68(1):28. [PMID: 40080284 DOI: 10.1007/s12016-025-09037-2]
- Ma Y, Zhang Z, Cao X, Guo D, Huang S, et al. Semaphorin 6A phase separation sustains a histone lactylation–dependent lactate buildup in pathological angiogenesis. Proc Natl Acad Sci U S A 2025;122(16):e2423677122. [PMID: 40244673 DOI: 10.1073/pnas.2423677122]
- Wang X, Huang J, Fan W, Li N, Yang H, et al. Lactate-binding protein DNMT3A in HRMECs promotes angiogenesis by upregulating VEGFA through HIF-1α lactylation. Genome Biol 2025;26(1):377. [PMID: 41168862 DOI: 10.1186/s13059-025-03845-7]
- Zhao M, Qian Y, He L, Peng T, Wang H, et al. Lactate-mediated histone lactylation promotes melanoma angiogenesis via IL-33/ST2 axis. Cell Death Dis 2025;16(1):701. [PMID: 41053006 DOI: 10.1038/s41419-025-08023-y]
- Zuo S, Li L, Tang J, Ye F, Sok, et al. Lactylation-boosted m5C RNA modification drives choroidal neovascularization. Research (Wash D C) 2025;8:0913. [PMID: 41049616 DOI: 10.34133/research.0913]
- Shen W, Nie Z, Wang M, Zhang H, Liu Y, et al. Monocyte-derived exosomal periostin driven by histone lactylation contributes to retinal neovascularization. Proc Natl Acad Sci U S A 2025;122(44):e2501704122. [PMID: 41150725 DOI: 10.1073/pnas.2501704122]
- Wang X, Fan W, Li N, Ma Y, Yao M, et al. YY1 lactylation in microglia promotes angiogenesis through transcription activation-mediated upregulation of FGF2. Genome Biol 2023;24(1):87. [PMID: 37085894 DOI: 10.1186/s13059-023-02931-y]
- Yu Z, Zhou X, Wang X. Metabolic reprogramming in hematologic malignancies: advances and clinical perspectives. Cancer Res 2022;82(17):2955-63. [PMID: 35771627 DOI: 10.1158/0008-5472.CAN-22-0917]
- Perez LM, Venugopal SV, Martin AS, Freedland SJ, Di Vizio D, et al. Mechanisms governing lineage plasticity and metabolic reprogramming in cancer. Trends Cancer 2024;10(11):1009-22. [PMID: 39218770 DOI: 10.1016/j.trecan.2024.08.001]
- Yang Y, Wu Y, Chen H, Xu Z, Lu R, et al. Research progress on the interaction between glucose metabolic reprogramming and lactylation in tumors. Front Immunol 2025;16:1595162. [PMID: 40755753 DOI: 10.3389/fimmu.2025.1595162]
- Wang T, Ye Z, Li Z, Jing DS, Fan GX, et al. Lactate–induced protein lactylation: a bridge between epigenetics and metabolic reprogramming in cancer. Cell Prolif 2023;56(10):e13478. [PMID: 37060186 DOI: 10.1111/cpr.13478]
- Zhang D, Liang C, Wu C, Hawanga M, Wan S, et al. Nonhistone lactylation: a hub for tumour metabolic reprogramming and epigenetic regulation. J Transl Med 2025;23(1):901. [PMID: 40797262 DOI: 10.1186/s12967-025-06813-8]
- Ge X, Zhu Y, Xiong J, Gu Y, Wang X, et al. Metabolic reprogramming through histone lactylation in microglia and macrophages recruits CD8+ T lymphocytes and aggravates spinal cord injury. Neuron 2025;113(14):2280-96.e8. [PMID: 40328251 DOI: 10.1016/j.neuron.2025.04.003]
- Chen L, Huang L, Gu Y, Cang W, Sun P, et al. Lactate-lactylation hands between metabolic reprogramming and immunosuppression. Int J Mol Sci 2022;23(19):11943. [PMID: 36233246 DOI: 10.3390/ijms231911943]
- Chen S, Qin T, Luo S, Wang F, Chen F, et al. Lactylation and viral infections: a novel link between metabolic reprogramming and immune regulation. PLoS Pathog 2025;21(7):e1013366. [PMID: 40720394 DOI: 10.1371/journal.ppat.1013366]
- She H, Hu Y, Zhao G, Du Y, Wu Y, et al. Dexmedetomidine ameliorates myocardial ischemia-reperfusion injury by inhibiting MDH2 lactylation via regulating metabolic reprogramming. Adv Sci (Weinh) 2024;11(48):e2409499. [PMID: 39467114 DOI: 10.1002/advs.202409499]
- Jia M, Yue X, Sun W, Zhou Q, Chang C, et al. ULK1-mediated metabolic reprogramming regulates Vps34 lipid kinase activity by its lactylation. Sci Adv 2023;9(22):eadg4993. [PMID: 37267363 DOI: 10.1126/sciadv.adg4993]
- Zheng J, She H, Han R, Tang J, Dou Y, et al. Dapk2 dysfunction leads to Mic60 lactylation and mitochondrial metabolic reprogramming, promoting lung cancer EGFR-TKI resistance and metastasis. Dev Cell 2025;60(23):3267-84.e7. [PMID: 40812308 DOI: 10.1016/j.devcel.2025.07.014]
- Yin YC, He J, Feng Y, Xu YY, Shi XH, et al. Filactate-induced lysine lactylation: a central node linking metabolic rewiring, epigenetic plasticity and therapeutic vulnerabilities in hepatocellular carcinoma. J Biochem Mol Toxicol 2025;39(12):e70622. [PMID: 41293912 DOI: 10.1002/jbt.70622]
- Liu X, Zhang Y, Li W, Zhou X. Lactylation, an emerging hallmark of metabolic reprogramming: current progress and open challenges. Front Cell Dev Biol 2022;10:972020. [PMID: 36092712 DOI: 10.3389/fcell.2022.972020]
- Chen AN, Luo Y, Yang YH, Fu JT, Geng XM, et al. Lactylation, a novel metabolic reprogramming code: current status and prospects. Front Immunol 2021;12:688910. [PMID: 34177945 DOI: 10.3389/fimmu.2021.688910]
- Dong Q, Zhang Q, Yang X, Nai S, Du X, et al. Glycolysis-stimulated Esrrb lactylation promotes the self-renewal and extraembryonic endoderm stem cell differentiation of embryonic stem cells. Int J Mol Sci 2024;25(5):2692. [PMID: 38473939 DOI: 10.3390/ijms25052692]
- Ye Z, Chen X, Zou J, Wu W, Yang J, et al. Lactylation’s role in bone health and disease: mechanistic insights and therapeutic potential. PeerJ 2025;13:e19534. [PMID: 40511385 DOI: 10.7717/peerj.19534]
- Chen Y, Wu J, Zhai L, Zhang T, Yin H, et al. Metabolic regulation of homologous recombination repair by MRE11 lactylation. Cell 2024;187(2):294-311.e21. [PMID: 38128537 DOI: 10.1016/j.cell.2023.11.022]
- Hagihara H, Shoji H, Otabi H, Toyoda A, Katoh K, et al. Protein lactylation induced by neural excitation. Cell Rep 2021;37(2):109820. [PMID: 34644564 DOI: 10.1016/j.celrep.2021.109820]
- Xu W, Zhao A, Han R, Wei J, Yu Q, et al. Extracellular lactate improves neurogenesis by modulating H3K9 lactylation and SnoN expression under hypoxic conditions. Stem Cell Res Ther 2025;16(1):462. [PMID: 40866997 DOI: 10.1186/s13287-025-04571-4]
- Wei L, Yang X, Wang J, Wang Z, Wang Q, et al. H3K18 lactylation of senescent microglia potentiates brain aging and Alzheimer’s disease through the NFκB signaling pathway. J Neuroinflammation 2023;20(1):208. [PMID: 37697347 DOI: 10.1186/s12974-023-02879-7]
- Zhou J, Zhang L, Peng J, Zhang X, Zhang F, et al. Astrocytic LRP1 enables mitochondria transfer to neurons and mitigates brain ischemic stroke by suppressing ARF1 lactylation. Cell Metab 2024;36(9):2054-68.e14. [PMID: 38906140 DOI: 10.1016/j.cmet.2024.05.016]
- Schafer MJ, White TA, Iijima K, Haak AJ, Ligresti G, et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 2017;8:14532. [PMID: 28230051 DOI: 10.1038/ncomms14532]
- Yanai H, Shteinberg A, Porat Z, Budovsky A, Braiman A, et al. Cellular senescence-like features of lung fibroblasts derived from idiopathic pulmonary fibrosis patients. Aging (Albany NY) 2015;7(9):664-72. [PMID: 26399448 DOI: 10.18632/aging.100807]
- Sun Z, Ji Z, He W, Duan R, Qu J, et al. Lactate accumulation induced by Akt2-PDK1 signaling promotes pulmonary fibrosis. FASEB J 2024;38(2):e23426. [PMID: 38226859 DOI: 10.1096/fj.202302063RR]
- Wu J, Li Y, Yang Y, Chen R, Wang H, et al. Wogonoside inhibits fibroblast activation and pulmonary fibrosis by dual regulation of Snail1 lactylation and PGC1α/PINK1-mediated mitophagy. Phytomedicine 2025;147:157240. [PMID: 40939548 DOI: 10.1016/j.phymed.2025.157240]
- Chen S, Wang Y, Zhang J, Liu B, Liu W, et al. YTHDC1 phase separation drives the nuclear export of m6A-modified lncNONMMUT062668.2 through the transport complex SRSF3-ALYREF-XPO5 to aggravate pulmonary fibrosis. Cell Death Dis 2025;16(1):279. [PMID: 40221424 DOI: 10.1038/s41419-025-07608-x]
- Wenhao L, Ling H, Wanqing F, Wanrong W, Chao C, et al. IGFBP7 and CCT2 are novel lactylation-driven mediators of endothelial-to-mesenchymal transition in idiopathic pulmonary fibrosis. Funct Integr Genomics 2025;25(1):250. [PMID: 41261179 DOI: 10.1007/s10142-025-01761-4]
- Xiong J, He J, Zhu J, Pan J, Liao W, et al. Lactylation-driven METTL3-mediated RNA m6A modification promotes immunosuppression of tumor-infiltrating myeloid cells. Mol Cell 2022;82(9):1660-77.e10. [PMID: 35320754 DOI: 10.1016/j.molcel.2022.02.033]
- Wu D, Spencer CB, Ortoga L, Zhang H, Miao C. Histone lactylation-regulated METTL3 promotes ferroptosis via m6A-modification on ACSL4 in sepsis-associated lung injury. Redox Biol 2024;74:103194. [PMID: 38852200 DOI: 10.1016/j.redox.2024.103194]
- Hahn EL, Halestrap AP, Gamelli RL. Expression of the lactate transporter MCT1 in macrophages. Shock 2000;13(4):253-60. [PMID: 10774612 DOI: 10.1097/00024382-200004000-00001]
- Brussow J, Feng K, Thiam F, Phogat S, Osei ET. Epithelial-fibroblast interactions in IPF: lessons from in vitro co-culture studies. Differentiation 2023;134:11-9. [PMID: 37738701 DOI: 10.1016/j.diff.2023.09.001]
- Komor AC, Kim YB, Packer MS, Zuris JA, Liu DR. Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 2016;533(7603):420-4. [PMID: 27096365 DOI: 10.1038/nature17946]
- Gaudelli NM, Komor AC, Rees HA, Packer MS, Badran AH, et al. Programmable base editing of A•T to G•C in Genomic DNA without DNA cleavage. Nature 2017;551(7681):464-71. [PMID: 29160308 DOI: 10.1038/nature24644]
- Chen PJ, Liu DR. Prime editing for precise and highly versatile genome manipulation. Nat Rev Genet 2023;24(3):161-77. [PMID: 36344749 DOI: 10.1038/s41576-022-00541-1]
- Sun Y, Chen Y, Peng T. A bioorthogonal chemical reporter for the detection and identification of protein lactylation. Chem Sci 2022;13(20):6019-27. [PMID: 35685793 DOI: 10.1039/d2sc00918h]
- David R, Richter MP, Beck-Sickinger AG. Expressed protein ligation. Method and applications. Eur J Biochem 2004;271(4):663-77. [PMID: 14764082 DOI: 10.1111/j.1432-1033.2004.03978.x]
- Currie MF, Singh SK, Ji M, Chatterjee C. The semisynthesis of site-specifically modified histones and histone-based probes of chromatin-modifying enzymes. Methods 2023;215:28-37. [PMID: 37244506 DOI: 10.1016/j.ymeth.2023.05.004]
- Kim CH, Kang M, Kim HJ, Chatterjee A, Schultz PG. Site-specific incorporation of ε-N-crotonyllysine into histones. Angew Chem Int Ed Engl 2012;51(29):7246-9. [PMID: 22689270 DOI: 10.1002/anie.201203349]
- Beckert S, Farrahi F, Aslam RS, Scheuenstuhl H, Königsrainer A, et al. Lactate stimulates endothelial cell migration. Wound Repair Regen 2006;14(3):321-4. [PMID: 16808811 DOI: 10.1111/j.1743-6109.2006.00127.x]
- Schenz J, Heilig L, Lohse T, Tichy L, Bomans K, et al. Extracellular lactate acts as a metabolic checkpoint and shapes monocyte function time dependently. Front Immunol 2021;12:729209. [PMID: 34899690 DOI: 10.3389/fimmu.2021.729209]
- Halestrap AP, Wilson MC. The monocarboxylate transporter family—role and regulation. IUBMB Life 2012;64(2):109-19. [PMID: 22162139 DOI: 10.1002/iub.572]
- Balasubramaniam S, Lewis B, Greed L, Meili D, Flier A, et al. Heterozygous monocarboxylate transporter 1 (MCT1, SLC16A1) deficiency as a cause of recurrent ketoacidosis. In: Morava E, Baumgartner M, Patterson M, Rahman S, Zschocke J, Peters V, editors. JIMD reports. Berlin, Heidelberg: Springer; 2016;29. pp. 33-8. [PMID: 26608392 DOI: 10.1007/8904_2015_519]
- McNeillis R, Greystoke A, Walton J, Bacon C, Keun H, et al. A case of malignant hyperlactaemic acidosis appearing upon treatment with the mono-carboxylase transporter 1 inhibitor AZD3965. Br J Cancer 2020;122(8):1141-5. [PMID: 32076124 DOI: 10.1038/s41416-020-0727-8]
- Katavolos P, Cain G, Farman C, Romero FA, Magnuson S, et al. Preclinical safety assessment of a highly selective and potent dual small-molecule inhibitor of CBP/P300 in rats and dogs. Toxicol Pathol 2020;48(3):465-80. [PMID: 32124659 DOI: 10.1177/0192623319898469]
- Matsuoka H, Fujimura T, Unami A, Yamada T, Noto T, et al. Novel method for selecting immunosuppressive histone deacetylase (HDAC) inhibitors with minimal thrombocytopenia. Biol Pharm Bull 2008;31(2):305-8. [PMID: 18239292 DOI: 10.1248/bpb.31.305]
- Duvic M, Vu J. Vorinostat in cutaneous T-cell lymphoma. Drugs Today (Barc) 2007;43(9):585-99. [PMID: 17940636 DOI: 10.1358/dot.2007.43.9.1112980]
- Osborne B, Bentley NL, Montgomery MK, Turner N. The role of mitochondrial sirtuins in health and disease. Free Radic Biol Med 2016;100:164-74. [PMID: 27164052 DOI: 10.1016/j.freeradbiomed.2016.04.197]
- Mourits VP, Helder LS, Matzaraki V, Koeken VACM, Groh L, et al. The role of sirtuin 1 on the induction of trained immunity. Cell Immunol 2021;366:104393. [PMID: 34147841 DOI: 10.1016/j.cellimm.2021.104393]
- King TE Jr, Bradford WZ, Castro-Bernardini S, Fagan EA, Glaspole I, et al. A phase 3 trial of pirfenidone in patients with idiopathic pulmonary fibrosis. N Engl J Med 2014;370(22):2083-92. [PMID: 24836312 DOI: 10.1056/NEJMoa1402582]
- Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, et al. Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med 2019;381(18):1718-27. [PMID: 31566307 DOI: 10.1056/NEJMoa1908681]
- Distler O, Highland KB, Gahlemann M, Azuma A, Fischer A, et al. Nintedanib for systemic sclerosis-associated interstitial lung disease. N Engl J Med 2019;380(26):2518-28. [PMID: 31112379 DOI: 10.1056/NEJMoa1903076]
- Vancheri C, Kreuter M, Richeldi L, Ryerson CJ, Valeyre D, et al. Nintedanib with add-on pirfenidone in idiopathic pulmonary fibrosis. Results of the INJOURNEY trial. Am J Respir Crit Care Med 2018;197(3):356-63. [PMID: 28889759 DOI: 10.1164/rccm.201706-1301OC]
- Flaherty KR, Fell CD, Huggins JT, Nunes H, Sussman R, et al. Safety of nintedanib added to pirfenidone treatment for idiopathic pulmonary fibrosis. Eur Respir J 2018;52(2):1800230. [PMID: 29946005 DOI: 10.1183/13993003.00230-2018]
- Thabut G, Crestani B. Treatments for idiopathic pulmonary fibrosis. N Engl J Med 2014;371(8):782. [PMID: 25140971 DOI: 10.1056/NEJMc1407776]