From Nociception to Regeneration: Temporal Reprogramming of Sensory Neurons in Skeletal Healing
1College & Hospital of Stomatology, Anhui Medical University, Anhui Provincial Key Laboratory of Oral Diseases Research, Hefei 230032, China
aThese authors contributed equally to this work.
*Correspondence to: Hengguo Zhang, College & Hospital of Stomatology, Anhui Medical University, Anhui Provincial Key Laboratory of Oral Diseases Research, Hefei 230032, Anhui, China, E-mail: zhanghengguo@ahmu.edu.cn
Received: March 2 2026; Revised: May 8 2026; Accepted: May 25 2026; Published Online: June 18 2026
Cite this paper:
Zhang Y, Wang H, Zhang H. From Nociception to Regeneration: Temporal Reprogramming of Sensory Neurons in Skeletal Healing. BIO Integration 2026; 7: 1–4.
DOI: 10.15212/bioi-2026-0037. Available at: https://bio-integration.org/
Download citation
© 2026 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Skeletal healing is constrained by a long-standing paradox. Specifically, analgesia is clinically essential, yet neural signaling is biologically required for effective repair. Recent evidence showed that bone-innervating sensory neurons are temporally plastic rather than functionally fixed. After injury, these neurons transition from an early nociceptive state to a later pro-regenerative secretory program, including trophic factors that are required for periosteal progenitor expansion and successful callus formation. Importantly, this switch is better interpreted as stage-linked than clock-like, broadly tracking the evolution from inflammatory injury signaling toward reparative callus formation with timing varying by skeletal site, age, and injury context. This dynamic framework helps explain why broad neural inhibition can relieve pain, while inadvertently compromising osteogenesis. The translational implication is not to reduce analgesia but to redesign analgesia. Future strategies should decouple nociceptive suppression from the loss of regenerative neural output. Defining therapeutic time windows and modality-specific effects on neurotrophic signaling as well as pain and developing biomarker-guided rescue strategies will be essential for next-generation skeletal management that preserves pain control and biological healing.
Keywords
Biomedical, engineering, neurosciences, orthopedics, physiology.
Mammalian skeletal healing is often characterized as intrinsically robust. However, in clinical practice skeletal healing is managed under a persistent therapeutic contradiction. Analgesia is necessary but neural activity appears biologically necessary as well. Standard pain-control strategies, including non-steroidal anti-inflammatory drugs (NSAIDs), local anesthetics, and regional blocks, are indispensable for patient comfort and function but standard pain-control strategies may also attenuate neural signals that support osteogenesis and callus maturation [1]. This long-standing tension has exposed a conceptual gap in skeletal healing. Indeed, pain and repair pathways have largely been treated as inseparable outputs of the same system.
Recent work in skeletal interoception has begun to close that conceptual gap. Sensory nerves are increasingly recognized not merely as nociceptive conduits, but as active regulators of mesenchymal stem-cell behavior and tissue homeostasis [2]. This regulation operates alongside a dynamic immune landscape, where early responders can exert opposing, osteo-inhibitory effects [3]. According to this view, the peripheral nervous system is part of the regenerative niche rather than an external observer of injury. What remains unresolved is whether this regulatory role is static (hard-wired by neuronal subtype) or dynamic (state-dependent after injury).
Xu et al. provided a decisive temporal framework for this question [4]. Methodologically, the authors utilized a robust combination of adeno-associated virus (AAV)-mediated retrograde anatomic mapping to identify bone-innervating sensory neurons, coupled with time-resolved single-cell transcriptomics (scRNA-seq) to capture dynamic neural state transitions. Xu et al. [4] used rigorous functional validation, including conditional neuronal FGF9 knockout and exogenous FGF9 rescue, to causally link these neural changes to skeletal repair. By combining these approaches, Xu et al. [4] showed that sensory neurons are not functionally fixed during skeletal repair. Instead, sensory neurons exhibit a phase-dependent state transition. Specifically, an early injury program enriched for nociceptive signaling is followed by a reparative program enriched for trophic output, including FGF9 (Figure 1). Conceptually, this trajectory aligns the early nociceptive state with the hematoma/inflammatory and early soft-callus phases of repair and the later trophic state with periosteal expansion, callus maturation, and bony bridging, although the exact timing is unlikely to be uniform across models, anatomic sites, ages, or species [5].
Figure 1 Temporal reprogramming of sensory neurons in skeletal healing. (A) Hyperexcitable neurons transmit nociceptive signals from the fracture site to the central nervous system. (B) Neurons transition to a regenerative phenotype. Released FGF9 promotes MSC differentiation into osteoblasts, forming a callus to bridge the defect.
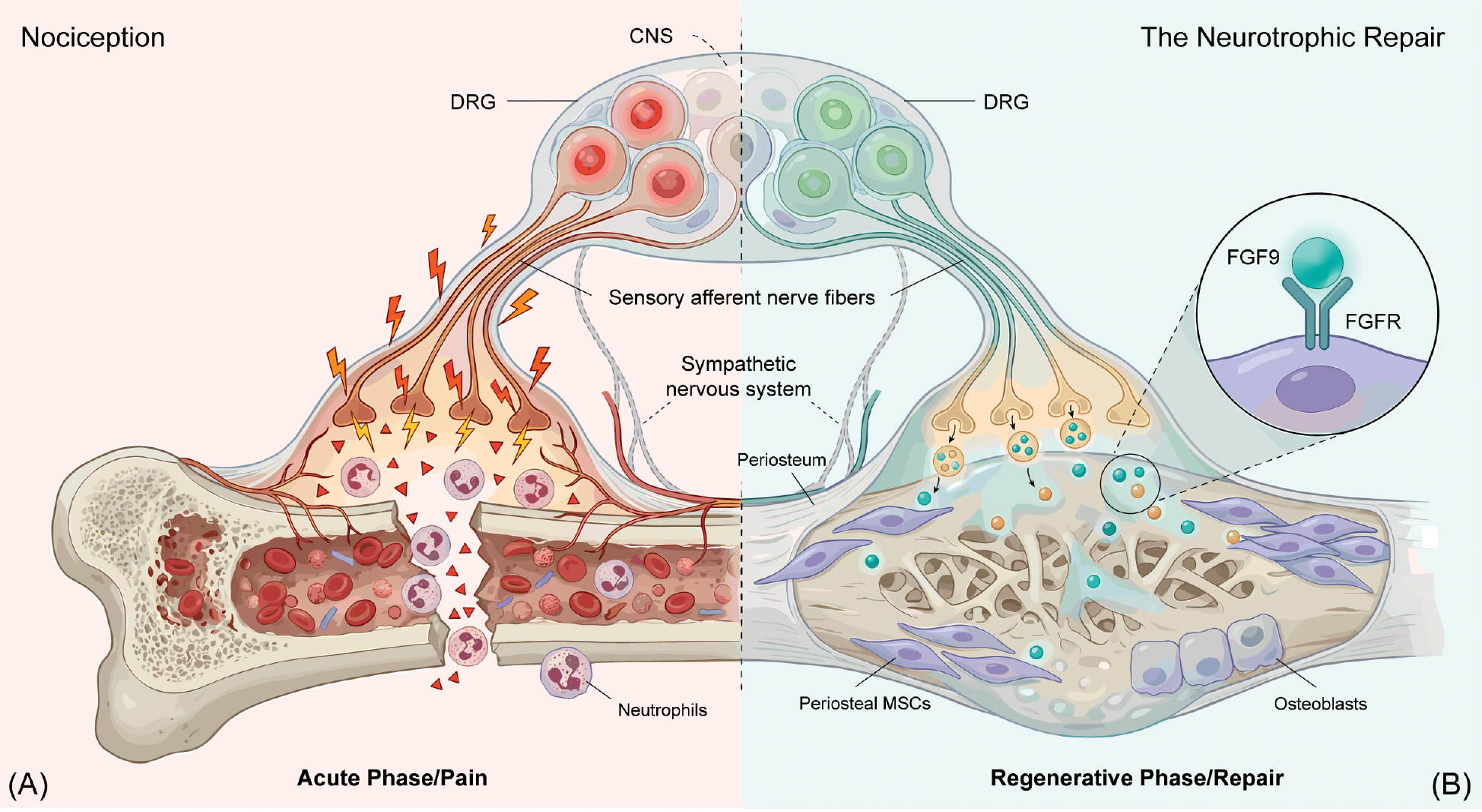
The importance of this study lies not only in descriptive resolution but in causal attribution. Xu et al. [4] identified a neuronal FGF9-dependent axis that is required for effective skeletal healing, linking neural state transitions to periosteal progenitor expansion and successful callus formation. Functional perturbation of neuronal FGF9 signaling supports a necessary role for this pathway in vivo and in loss-of-function contexts, repair fails toward an atrophic non-union phenotype, indicating that nerve-derived trophic signaling is not epiphenomenal but functionally required. At the same time, FGF9 is best viewed as a mechanistic anchor rather than an exclusive effector. Reparative sensory neurons deploy a broader pro-regenerative secretome and parallel outputs that remain to be resolved. Conceptually, the nervous system emerges as a two-stage regulator of repair (an acute protective module and a delayed anabolic module).
This temporal model extends prior human sensory-neuron atlases in a productive way. Spatial transcriptomic mapping of human dorsal root ganglia (DRG) has defined nociceptor classes with extraordinary precision but such atlases are necessarily snapshots [6]. Xu et al. [4] added the missing variable of time under injury conditions, demonstrating that reparative biology depends not only on neuronal identity but also on neuronal trajectory. This is a critical distinction for translational science. In fact, a static taxonomy alone cannot predict time-sensitive therapeutic windows.
The clinical implications are immediate. If bone-innervating sensory neurons are a required source of pro-regenerative cues, then “broad neural silencing” becomes a biologically blunt strategy. The goal should not be less analgesia but smarter analgesia. Therapeutic interventions should aim to suppress nociceptive transmission while preserving trophic signaling, strategically timing treatments to minimize disruption of neuroprotective pathways, or substituting alternative sources of trophic support when necessary. Importantly, NSAIDs, local anesthetics, regional nerve blocks, opioids, and neuromodulatory approaches should not be assumed to be biologically interchangeable. NSAIDs, local anesthetics, regional nerve blocks, opioids, and neuromodulatory approaches may differ in how neuronal excitability, local afferent activity, and downstream trophic transcriptional programs are affected, even when pain scores appear similar. For example, NSAIDs may interfere with prostaglandin-mediated osteogenesis, while local anesthetics primarily suppress afferent excitability. Opioids may exert systemic endocrine effects that indirectly influence bone metabolism [7, 8]. Accordingly, future comparisons should evaluate pain relief and the effects on regenerative signaling, callus formation, radiographic union, and functional recovery. Candidate biomarkers may include circulating or local neurotrophic factors (e.g., FGF9, NGF, and CGRP), assessed in phase-aligned sampling windows to capture the temporal dynamics of neuronal state transitions. Xu et al. [4] therefore shifted the therapeutic objective from global inhibition toward functional decoupling. In practical terms, this shift suggests the following three translational priorities:
- Define when the nociceptive-to-regenerative transition occurs across skeletal contexts, genders, and age groups, and relate the nociceptive-to-regenerative transition to canonical phases of fracture healing rather than to a single universal timeline.
- Compare analgesic modalities by the differential impact on neurotrophic programs, callus formation, radiographic union, and functional recovery, rather than by pain scores alone.
- Test whether pathway-selective augmentation (for example, local pro-regenerative cue support or biomaterial-enabled trophic delivery) can rescue healing when neural suppression is unavoidable [9].
Several caveats remain. The strongest mechanistic evidence is preclinical and skeletal biology varies by age, anatomical site, loading environment, and comorbidity. The foundational study conducted by Xu et al. [4] primarily utilized male mice, leaving the role of sexual dimorphism in neural-skeletal crosstalk unaddressed. A significant translational gap exists given established gender differences in pain processing and bone metabolism [10]. Furthermore, age-related dysregulation of neurotrophin signaling, such as aberrant NGF expression dynamics, may exacerbate the deleterious effects of neural inhibition in elderly fracture patients [11]. The direct translation of these findings should be interpreted with caution and based on documented species differences in human DRG diversity, as well as the yet-unaddressed contributions of the sympathetic and autonomic nervous systems, which are also known to modulate osteoblast and osteoclast activity in the healing niche [5, 6]. Moreover, while FGF9 represents a critical necessary factor, FGF9 is not likely to be the sole trophic output of reparative neurons and operates alongside co-secreted factors, such as Sonic Hedgehog, to drive vital neuroimmune and neurovascular congruence. For example, sensory neuron–derived signals may coordinate angiogenesis and immune-cell recruitment, processes that are tightly coupled to callus formation and remodeling [12]. The regulation of progenitor fate likely involves intricate intracellular coordination, such as nuclear FGF2-mediated chromatin remodeling [13]. But these limitations do not weaken the central advance, rather define the next experiments. The key contribution of Xu et al. [4] is to make the neural contribution to skeletal healing experimentally tractable in space, time, and mechanism.
Skeletal healing has long assumed that pain control and biological repair exist in zero-sum opposition. That assumption is now less defensible. Sensory neurons are not simply signals to block. Sensory neurons are state-dependent partners in regeneration. By mapping and functionally validating this transition, Xu et al. [4] offered a mechanistic blueprint for a new clinical principle, i.e., treat pain without extinguishing the neural biology of healing.
Author contributions
Y.Z., H.W., and H.Z. wrote the manuscript. All authors have read and approved the article.
Funding
This work was supported by grants from the National Natural Science Foundation of China (nos. 82201026 & 82571071), the Open Research project of State Key Laboratory of Oral Diseases (no. SKLOD2024OF03), and the Anhui Province Outstanding Young Teachers Development Program (no. YQYB2024013).
Acknowledgments
We thank the members of our group for helpful discussions. Figure 1 was created by using Biorender (https://www.biorender.com/), released under a Creative Commons Attribution-NonCommercial-NoDerivs 4.0 International license.
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Wheatley BM, Nappo KE, Christensen DL, Holman AM, Brooks DI, et al. Effect of NSAIDs on bone healing rates: a meta-analysis. J Am Acad Orthop Surg 2019;27(7):e330-e6. [PMID: 30260913 DOI: 10.5435/JAAOS-D-17-00727]
- Hu B, Lv X, Chen H, Xue P, Gao B, et al. Sensory nerves regulate mesenchymal stromal cell lineage commitment by tuning sympathetic tones. J Clin Invest 2020;130(7):3483-98. [PMID: 32191640 DOI: 10.1172/JCI131554]
- Lin W, Li Q, Liu L, Wang Q, Zhang D, et al. Early infiltrating NKT lymphocytes attenuate bone regeneration through secretion of CXCL2. Sci Adv 2024;10(20):eadl6343. [PMID: 38758783 DOI: 10.1126/sciadv.adl6343]
- Xu M, Li Z, Thottappillil N, Cherief M, Zhu M, et al. Mapping somatosensory afferent circuitry to bone identifies neurotrophic signals required for fracture healing. Science 2026;391(6781):eadr9608. [PMID: 41505527 DOI: 10.1126/science.adr9608]
- Parker RS, Nazzal MK, Morris AJ, Fehrenbacher JC, White FA, et al. Role of the neurologic system in fracture healing: an extensive review. Curr Osteoporos Rep 2024;22(1):205-16. [PMID: 38236509 DOI: 10.1007/s11914-023-00844-0]
- Tavares-Ferreira D, Shiers S, Ray PR, Wangzhou A, Jeevakumar V, et al. Spatial transcriptomics of dorsal root ganglia identifies molecular signatures of human nociceptors. Sci Transl Med 2022;14(632):eabj8186. [PMID: 35171654 DOI: 10.1126/scitranslmed.abj8186]
- Coluzzi F, Scerpa MS, Centanni M. The effect of opiates on bone formation and bone healing. Curr Osteoporos Rep 2020;18(3):325-35. [PMID: 32249381 DOI: 10.1007/s11914-020-00585-4]
- Lisowska B, Kosson D, Domaracka K. Positives and negatives of nonsteroidal anti-inflammatory drugs in bone healing: the effects of these drugs on bone repair. Drug Des Devel Ther 2018;12:1809-14. [PMID: 29950815 DOI: 10.2147/DDDT.S164565]
- Venkatesan J, Liu W, Madry H, Cucchiarini M. Alginate hydrogel-guided rAAV-mediated FGF-2 and TGF-β delivery and overexpression stimulates the biological activities of human meniscal fibrochondrocytes for meniscus repair. Eur Cells Mater 2024;47:1-14. [DOI: 10.22203/eCM.v047a01]
- Mogil JS. Qualitative sex differences in pain processing: emerging evidence of a biased literature. Nat Rev Neurosci 2020;21(7):353-65. [PMID: 32440016 DOI: 10.1038/s41583-020-0310-6]
- Sekiguchi H, Inoue G, Shoji S, Tazawa R, Kuroda A, et al. Expression of nerve growth factor in the callus during fracture healing in a fracture model in aged mice. Biomed Mater Eng 2022;33(2):131-137. [PMID: 34487017 DOI: 10.3233/BME-211284]
- Lu YZ, Nayer B, Singh SK, Alshoubaki YK, Yuan E, et al. CGRP sensory neurons promote tissue healing via neutrophils and macrophages. Nature 2024;628(8008):604-11. [PMID: 38538784 DOI: 10.1038/s41586-024-07237-y]
- Zhang H, Wang Z, Liu Z, Li X, Sun W, et al. Nuclear FGF2 orchestrates phase separation-mediated rDNA chromatin architecture to control BMSCs cell fate. Bone Res 2025;13(1):80. [PMID: 40993129 DOI: 10.1038/s41413-025-00451-y]