Large-Scale Chemical Space Exploration Identifies DE19725241 as a Candidate Fat Mass and Obesity-Associated Protein-Targeting Compound with Selective Activity in Pancreatic Cancer Cells
1Zhejiang University School of Medicine Sir Run Run Shaw Hospital, Department of General Surgery, Hangzhou, China
2School of Rare Earths, University of Science and Technology of China, Hefei, China
3Jiangnan University, Wuxi, China
4Zhejiang University School of Medicine Sir Run Run Shaw Hospital, Department of Thoracic Surgery, Hangzhou, China
5Zhejiang Provincial People’s Hospital, Hangzhou, China
6Department of Colorectal Surgery, Sir Run Run Shaw Hospital, Zhejiang University School of Medicine, Hangzhou, China
7Chinese Academy of Sciences, Jiangxi Institute of Rare Earths, Ganzhou, China
*Correspondence to: Fangyu Ren, Fundamental Science Center of Rare Earths, Ganjiang Innovation Academy, Chinese Academy of Sciences, Ganzhou, Jiangxi, China, E-mail: fangyu.ren@gia.cas.cn
Received: December 24 2025; Revised: March 22 2026; Accepted: May 14 2026; Published Online: July 6 2026
Cite this paper:
Xu C, Cai Z, Wang D et al. Large-Scale Chemical Space Exploration Identifies DE19725241 as a Candidate Fat Mass and Obesity-Associated Protein-Targeting Compound with Selective Activity in Pancreatic Cancer Cells. BIO Integration 2026; 7: 1–17.
DOI: 10.15212/bioi-2025-0225. Available at: https://bio-integration.org/
Download citation
© 2026 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Background: Fat mass and obesity-associated protein (FTO), an epitranscriptomic regulator, has been implicated in cancer progression and immune regulation but the therapeutic relevance in pancreatic cancer is unclear.
Methods: TCGA and GTEx transcriptomic datasets were analyzed to evaluate FTO expression and prognostic associations in pancreatic cancer. Candidate FTO-targeting compounds were identified by active learning-assisted virtual screening of > 22 million compounds. The lead candidate, DE19725241, was further assessed by binding pose metadynamics, molecular dynamics simulations in multiple environments, MM/GBSA calculations, and in vitro testing in three pancreatic cancer cell lines and one normal pancreatic epithelial cell line.
Results: FTO was overexpressed in pancreatic tumors and associated with poorer overall survival. DE19725241 showed favorable predicted interactions with FTO, particularly with ARG-96, TYR-108, and GLU-234, and exhibited moderate but selective antiproliferative activity in pancreatic cancer cells.
Conclusions: DE19725241 represents a potential early-stage scaffold for developing FTO-targeted strategies in pancreatic cancer.
Keywords
Active learning-assisted virtual screening, ADMET, CCK-8, chemical space library, chemistry, FTO-targeting compound, medicinal < chemistry, molecular dynamics simulation, pancreatic cancer, transcriptomic analysis
Introduction
Pancreatic ductal adenocarcinoma (PDAC) has now replaced breast cancer as the third most common cause of cancer-related deaths in the United States [1] with projections indicating that PDAC will replace colorectal cancer to become the second deadliest cancer after lung cancer by 2040. The high PDAC mortality rate is primarily due to late-stage detection, in which the cancer has already spread to distant organs [2, 3]. Aside from a small fraction (10%–15%) of cases linked to inherited mutations or established risk factors, such as mucinous cystic neoplasms and chronic inflammation of the pancreas, the majority of PDAC cases lack a clear causative factor [4]. The challenge of early diagnosis is further exacerbated by the silent nature of the disease in the localized form or non-specific symptoms, the scarcity of effective early-stage tumor markers, and the deep abdominal location of the pancreas, which hinders easy screening in a regular clinical setting [5]. The increasing prevalence of PDAC, which is fueled by factors, such as the obesity crisis and increased average lifespan, highlights the critical and immediate need for innovative treatment approaches that can offer significant benefits to most patients with PDAC.
The treatment of pancreatic cancer involves various molecular targeted drugs focusing on targets, such as the epidermal growth factor receptor [EGFR] [6], KRAS protein [7], cyclin-dependent kinases [CDKs] [8], and human epidermal growth factor receptor 2 [HER2] [9]. While drugs, like gefitinib [10, 11] and erlotinib [12], that target EGFR can inhibit tumor growth, such drugs may cause severe skin rashes and digestive system reactions. KRAS mutations [13], which are common in pancreatic cancer, pose challenges in drug development due to potential widespread cytotoxicity that limits application. CDK inhibitors, like palbociclib [14], block tumor cell proliferation but may cause hematologic toxicity and liver function abnormalities [15]. In addition, researchers are exploring inhibitors of the PI3K/Akt/mTOR pathway, which are crucial for cell survival and proliferation in various cancers, including pancreatic cancer [16]. Despite the efficacy, PI3K/Akt/mTOR pathway inhibitors can induce significant side effects, such as hyperglycemia and immunosuppression, which post risks to overall patient health [17]. These complexities underscore the necessity for developing new therapeutic targets, like fat mass and obesity-associated protein, which offers a potentially safer and more effective treatment avenue by regulating N6-methyladenosine (m6A) RNA modifications affecting cancer-related gene expression [18].
Among the extensive range of modified nucleotides within RNA, m6A has emerged as the most common and significant modification in the mRNAs of eukaryotes [19]. The discovery of FTO as the initial enzyme capable of demethylating m6A in RNA via α-ketoglutarate (α-KG)- and Fe(II)-dependent [20] processes revealed that m6A modification is a dynamic and reversible process [21]. Recent findings highlight the elevated expression and pivotal involvement of FTO, a m6A demethylase, in leukemia [22]. A corollary study demonstrated that FTO is vulnerable to inhibition by R-2-hydroxyglutarate (R-2HG), which subsequently exhibits inherent antileukemic properties through downregulation of FTO [23]. Moreover, numerous studies have revealed the excessive expression and potential tumorigenic influence of FTO across a variety of solid cancer types [24]. Together, these insights position FTO as a valuable target for cancer therapy, underscoring the need for an in-depth exploration of the role of FTO in cancer processes and the pursuit of effective FTO-focused therapeutic strategies.
A range of molecules have been identified as potential FTO inhibitors in various studies, including well-documented compounds in DrugBank, such as rhein [25], N-CDPCB [26], meclofenamic acid [MA] [27], MO-I-500 [28], fluorescein [29], and R-2HG [23]. Despite the identification, these agents exhibit limited clinical applicability because of the relatively weak biological activity and lack precision and sensitivity [30]. Recent advances in the development of FTO inhibitors have revealed several additional promising compounds. Diacerein [31], which was identified through an innovative quantum-dot-based FRET sensor technique, selectively inhibits FTO activity without the need for structural cofactor mimicry. In addition, compounds from the oxetanyl class, derived from previously identified compounds, have demonstrated impressive FTO inhibition. Notably, FTO-43, a member of this class, has shown nanomolar potency and robust antiproliferative effects across various cancer models. This compound exemplifies the potential of oxetanyl derivatives for effectively targeting FTO to combat malignancies [32]. Moreover, small-molecule inhibitors developed through comprehensive high-throughput screening have been shown to potently inhibit FTO, underscoring the potential therapeutic applications in treating neurodegenerative diseases [33]. Finally, pioneering research has led to the development of FTO inhibitor-loaded GSH-bioimprinted nanocomposites [GNPIPP12MA] [22]. These nanocomposites target leukemia stem cells and induce ferroptosis, significantly enhancing the efficacy of immune checkpoint inhibitors in leukemia treatment. In addition, MA derivatives (FB23 and FB23-2) have shown superior ability to decrease FTO function and increase the survival rates of cells afflicted by acute myeloid leukemia (AML). However, the effectiveness, as measured by the concentration required to reduce AML cell survival by one-half (IC50), remains suboptimal, exceeding 1 μM for FB23-2 and 20 μM for FB23 [30]. Although FB23-2 has demonstrated a significant slowing effect on AML progression in murine models, which serves as a concrete example of the potential benefits of FTO pharmacologic intervention in AML treatment, the extent of inhibition achieved falls short of expectations. Consequently, the discovery and development of more potent FTO inhibitors are urgently needed to generate more effective therapeutic strategies for AML and other cancer types, including pancreatic cancer.
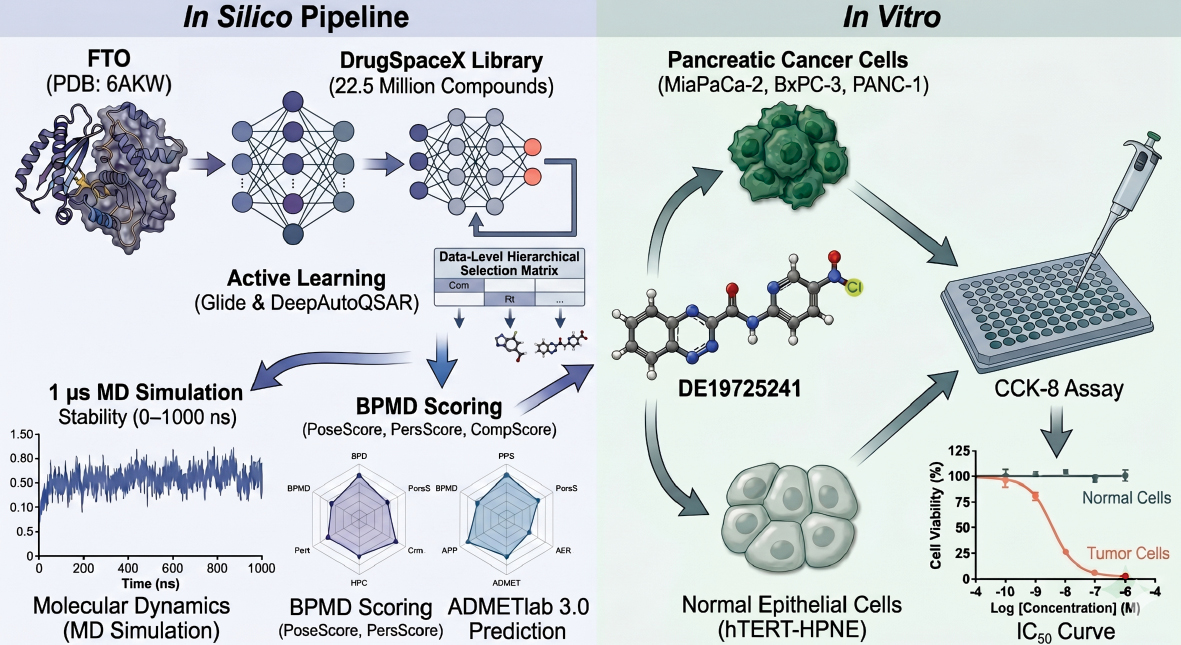
To further explore and discover effective inhibitors targeting FTO, an innovative screening strategy was adopted herein to identify potential candidate compounds from the DrugSpaceX Drug-like Subset [34], which contains 22,556,593 compounds. An active learning model [35], docking score, MMGBSA [36], state penalty, and ligand strain energy [37] calculations were combined with various scoring methods for preliminary screening. Subsequently, interaction analysis, binding pose metadynamics [BPMD] [38], and classical molecular dynamic simulations of varying durations were used to assess the binding stability and environmental adaptability of the compounds, ultimately evaluating the viability as drugs through ADMET predictions. This screening strategy was designed to yield strong candidate compounds for subsequent experimental research and drug development. The workflow of the current study is illustrated in Figure 1.
Figure 1 Research flow chart.
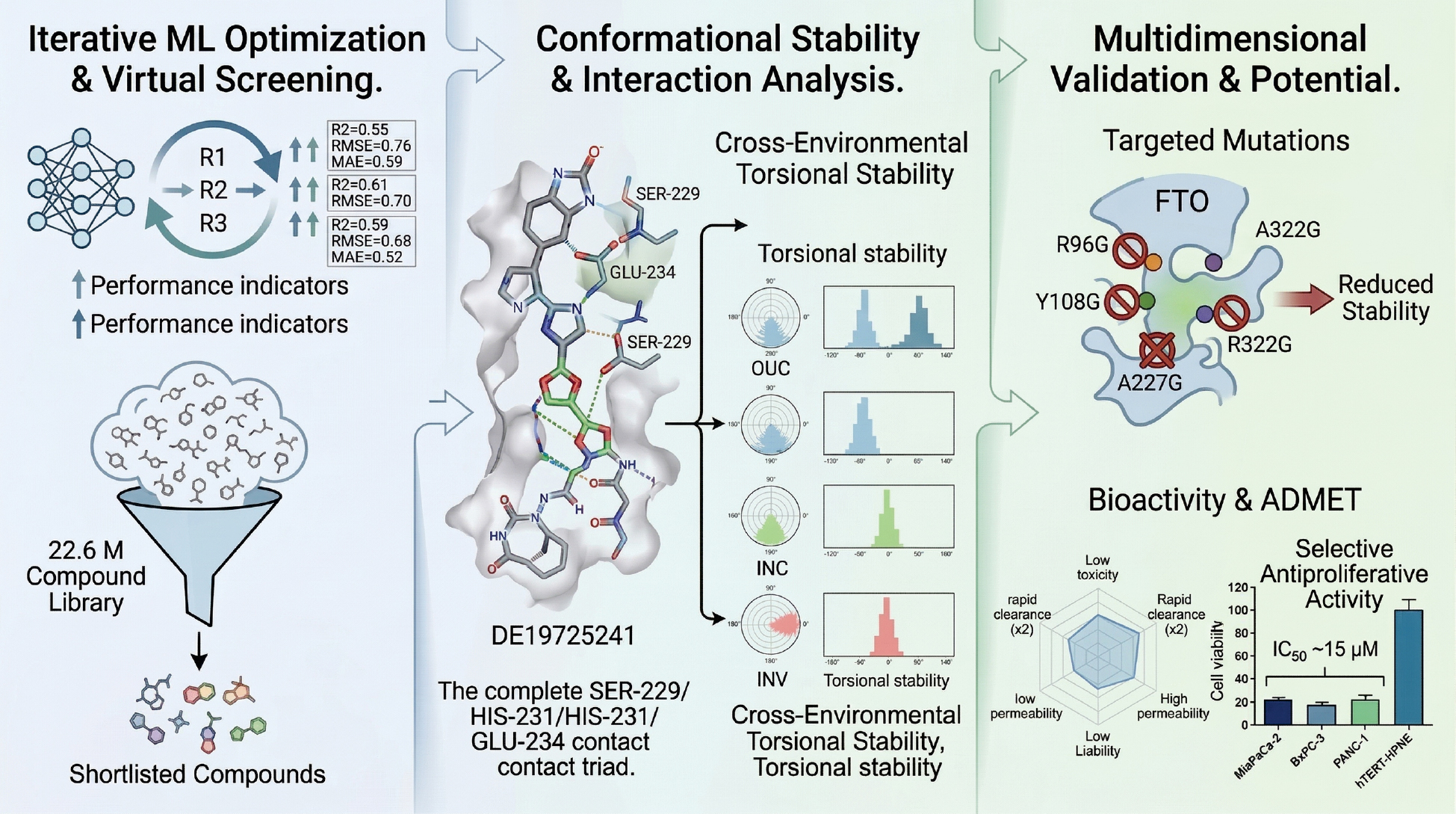
Methods
Protein preparation
Structure-guided computational techniques were applied with the FTO crystal structure [PDB ID: 6AKW] [30] chosen as the receptor template in the search for inhibitors targeting FTO. The Schrödinger suite protein preparation wizard (“Schrödinger Release 2023–3: protein preparation,” 2023) was used to refine the complex for the simulations. Specifically, the addition of absent hydrogen atoms, amendment of the charge states of the metal ions, bond order assignments in the hetero atoms (HET_ groups, assessment of ligand protonation and related energetic costs, adjustment of the protonation of the histidine residues, correction of possible misplacements of heavy atoms, enhancement of the protein’s hydrogen bonding structure, and execution of constrained energy minimization were implemented. The specific site on the 3D structure of the receptor where FB23 interacts was identified as the focal point for ligand screening, leading to the establishment of a designated grid for this purpose.
Active learning-based virtual screening
Active Learning Glide, an advanced docking workflow designed for ultra-large virtual screening, was used to evaluate the DrugSpaceX (https://drugspacex.simm.ac.cn/) Drug-like Subset with Glide SP [39]. Rather than explicitly docking all 22,556,593 compounds, the workflow docked 50,000 compounds per round for 3 rounds (150,000 compounds in total, approximately 0.66% of the full library) during the model-building stage. The resulting Glide scores served as physics-based labels for training the machine learning model and model performance was monitored using standard regression metrics, including the root mean square error (RMSE), coefficient of determination (R2), and mean absolute error (MAE). Following three iterative training cycles, the optimized model was applied to the remaining library to prioritize candidates for downstream selection and rescoring. In this way, the active learning framework was used as an efficiency-oriented prioritization strategy that retained docking-based ranking information, while markedly reducing the number of explicit docking calculations required for ultra-large screening. This workflow was implemented using Schrödinger’s Active Learning Glide and DeepAutoQSAR platform (“Schrödinger Release 2023-3: DeepAutoQSAR,” 2023).
After completion of the iterative training, the optimized model was applied to the entire DrugSpaceX library to prioritize candidate ligands for conventional Glide SP redocking and more precise pose evaluation. Subsequent prioritization was based on an integrated assessment of the docking score, state penalty, ligand strain energy, molecular mechanics generalized born surface area (MMGBSA) binding free energy, and binding-mode inspection. Specifically, compounds were retained only if the compounds simultaneously met the following thresholds, followed by manual inspection of docking poses: Glide SP docking score < −9.0; State Penalty = 0; Ligand Strain Energy < 1.0 kcal/mol; and MMGBSA dG Bind < −50 kcal/mol. In the present study the active learning workflow was used as an efficiency-oriented prioritization strategy rather than a formal benchmarking exercise. A direct benchmark against exhaustive Glide docking of the full 22.6-million-compound library was not performed because such a brute-force screen was beyond the computational scope of this work.
Binding pose metadynamics
A hill height setting of 0.05 kcal/mol and a hill width of 0.02 Å were used for BPMD [40]. The calculation of the RMSD was based on a 3 Å threshold for the distance between protein residues and ligands [41]. The initial steps included arranging the system in a simple point change (SPC) water model, followed by reducing the system energy through minimization, and applying necessary constraints with an incremental increase in the temperature to 300 K. Then, metadynamics were considered. The reference for the metadynamics approach was established by the final 0.5 nanoseconds from a non-biased molecular dynamics (MD) simulation.
Three BPMD scores were used to evaluate ligand binding stability: PoseScore; PersScore; and CompScore. PoseScore measures the mean RMSD deviation of the ligand from the starting configuration with rapid increases indicating possible binding instability. A PoseScore < 2 Å was considered indicative of a stable ligand-protein interaction [38]. PersScore evaluates the persistence of hydrogen-bond interactions throughout the simulation with higher values indicating greater binding fidelity. CompScore combines PoseScore and PersScore into a composite metric with lower values suggesting a more stable ligand-protein complex [38].
Molecular dynamics simulations with desmond
MD simulations of the FTO-compound interactions were performed to evaluate the stability of the compounds over time. An orthorhombic simulation box was configured, which ensured a minimum distance of 10 Å from any point on the box to the nearest point of the FTO-compound complex. The system was immersed in a TIP3P explicit solvent and charge neutrality was achieved by adding appropriate counterions. The solvent included 150 mM NaCl or KCl to replicate physiologic conditions. The energy of the setup was minimized by adjusting the OPLS2005 force field until the energy gradient reached 1 kcal mol−1Å−1. Afterwards, MD simulations of the system were conducted for 100 or 1000 ns. A constant temperature (300 K or 310 K) and pressure (atmospheric levels) were maintained with a Nose‒Hoover Chain thermostat and Martyna‒Tobias‒Klein barostat, respectively, with default relaxation settings activated prior to the start of the simulation.
ADMET prediction
The leading candidates in the structure-oriented virtual screening and MD simulations were identified and subsequently evaluated with ADMETlab 3.0 [42] [https://admetlab3.scbdd.com/]. The aim of this evaluation was to elucidate the pharmacokinetic profiles, bioavailability, and general appropriateness of these compounds for potential therapeutic use.
Cell viability assay
The anti-proliferative activity of DE19725241 was assessed using the Cell Counting Kit-8 [CCK-8] Dojindo, Kumamoto, Japan) according to the manufacturer’s instructions. The following 4 cell lines were used in this study: MIA PaCa-2, BxPC-3, PANC-1, and hTERT-HPNE cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were cultured under standard conditions (37 °C and 5% CO2) in the recommended culture media supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin.
Cells were seeded in 96-well plates at a density of 5 × 103 cells per well and allowed to adhere overnight. DE19725241 was then added at various concentrations (0, 3.125, 6.25, 12.5, 25, 50, and 100 μM) with 6 replicates for each concentration. After 48 h of treatment, 10 μL of CCK-8 reagent was added to each well and incubated for 2 h at 37 °C. Absorbance was measured at 450 nm using a microplate reader (BioTek, city, state, USA).
Cell viability was expressed as a percentage relative to the untreated control group. IC50 values were calculated using non-linear regression analysis with GraphPad Prism 9. The selectivity of DE19725241 was evaluated by comparing the inhibitory effects in pancreatic cancer and normal pancreatic epithelial cells.
Results
Iterative optimization of the machine learning model enhances predictive accuracy
In this study the predictive performance of the machine learning model was progressively optimized over three consecutive iterations. The active learning workflow refined the relationship between explicitly docked compounds and model-predicted scores at each stage, thereby improving the prioritization of compounds for subsequent screening. Model performance was evaluated using standard regression metrics, including the R2, RMSE, and MAE, which together reflect both predictive accuracy and the deviation between predicted and actual values, as summarized in Figure 2A-C and Table 1. From a screening-efficiency perspective, only 150,000 compounds were explicitly docked during model training before the optimized model was used to prioritize the remaining 22.6-million-compound library.
Figure 2 Training process of the active learning model and representative selection outcome. Three iterations of active learning model training are shown: Round 1 (R1; A), Round 2 (R2; B), and Round 3 (R3; C). Following a selection process incorporating multiple scoring criteria and manual review, 14 compounds were retained. Representative 2D structures are shown alongside FB23, with pink regions highlighting structural similarities to the reference scaffold (D).
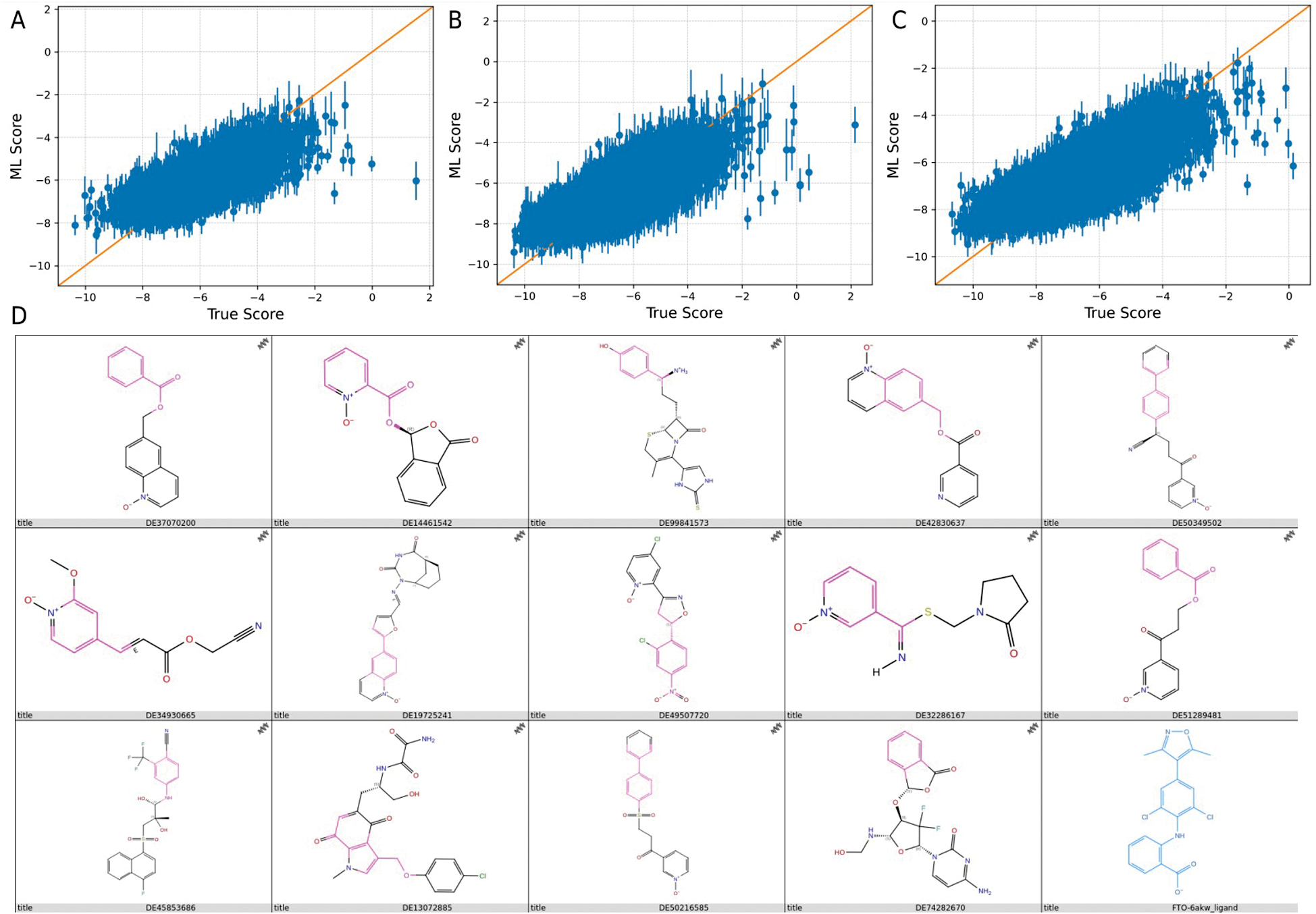
Table 1 The Evaluation Score of the Active Learning Model Over Three Iterations
| Iteration | R2 | RMSE | MAE |
|---|---|---|---|
| R1 | 0.55 | 0.76 | 0.59 |
| R2 | 0.61 | 0.7 | 0.54 |
| R3 | 0.59 | 0.68 | 0.52 |
The model yielded an R2 of 0.55 in the first iteration (R1), indicating that 55% of the variance in the data could be explained by the model. However, the relatively high RMSE (0.76) and MAE (0.59) values suggested noticeable discrepancies between predicted and observed outputs.
The model exhibited enhanced performance during the second iteration (R2). The R2 increased to 0.61, reflecting improved explanatory power and predictive accuracy. Concurrently, the RMSE decreased to 0.70 and the MAE decreased to 0.54, indicating reduced prediction error and enhanced consistency between predicted and actual values.
The third iteration (R3) led to further refinement. Although the R2 value slightly declined to 0.59 compared to the second iteration, the R2 value remained above the initial value in R1, suggesting sustained model interpretability. Importantly, the RMSE and MAE continued to decrease, reaching 0.68 and 0.52, respectively, further validating incremental improvements in predictive precision.
Collectively, these results demonstrated that the model benefited from iterative optimization with each round contributing to a reduction in prediction errors and improved predictive robustness. In practical terms, the active learning workflow enabled prospective prioritization of a 22.6-million-compound library after explicitly docking only a small fraction of compounds during model training.
Identifying promising candidates through iterative machine learning analysis
Following completion of model training, the active learning framework was applied to screen the DrugSpaceX drug-like subset, comprising 22,556,593 compounds. Hit selection was performed using a stepwise filtering strategy rather than a single composite score. Specifically, compounds were retained only if the compounds simultaneously met the following criteria: Glide SP docking score < −9.0; State Penalty = 0; Ligand Strain Energy < 1.0 kcal/mol; and MMGBSA dG Bind < −50 kcal/mol. These thresholds were used to prioritize compounds with favorable predicted binding, no additional binding-state penalty, limited conformational strain, and consistently favorable binding free energy.
Compounds satisfying all four criteria were then subjected to manual inspection of docking poses to remove candidates with unfavorable binding orientations or limited occupancy of the FTO substrate-binding pocket. Using this combined score- and pose-based selection procedure, 14 structurally diverse compounds were retained as preliminary hits (Table 2; Figure 2D).
Table 2 Results After Screening with the Active Learning Model and the Corresponding Docking Scores
| Compound ID | Docking Score | State Penalty | Lig Strain Energy | MMGBSA dG Bind |
|---|---|---|---|---|
| DE37070200 | −9.848 | 0 | 0.853 | −60.27 |
| DE14461542 | −9.788 | 0 | 0.938 | −51.13 |
| DE99841573 | −9.773 | 0 | 0.984 | −64.91 |
| DE42830637 | −9.758 | 0 | 0.84 | −61.07 |
| DE50349502 | −9.553 | 0 | 0.732 | −62.27 |
| DE34930665 | −9.407 | 0 | 0.93 | −53.18 |
| DE19725241 | −9.352 | 0 | 0.679 | −63.13 |
| DE49507720 | −9.256 | 0 | 0.994 | −54.33 |
| DE32286167 | −9.141 | 0 | 0.996 | −50.19 |
| DE51289481 | −9.127 | 0 | 0.704 | −59.48 |
| DE45853686 | −9.115 | 0 | 0.269 | −52.88 |
| DE13072885 | −9.109 | 0 | 0.766 | −72.09 |
| DE50216585 | −9.098 | 0 | 0.853 | −52.8 |
| DE74282670 | −9.043 | 0 | 0.421 | −60.7 |
To further refine the candidate set, the planar interaction diagrams in Figure 3 were explicitly examined to determine whether the retained ligands reproduced recurring contact features within the FTO binding pocket. A clear interaction pattern centered on SER-229 was observed in 13 of the 14 compounds, while GLU-234 and HIS-231 were also frequently involved, appearing in 10 and 8 compounds, respectively. Notably, six compounds (DE37070200, DE99841573, DE42830637, DE19725241, DE49507720, and DE45853686) shared the complete SER-229/HIS-231/GLU-234 contact triad, suggesting a common and favorable binding orientation. Additional candidates reproduced partial but still informative patterns, as follows: DE14461542 engaged SER-229 and HIS-231; DE32286167 and DE51289481 engaged SER-229 and GLU-234; DE34930665 formed contacts with ARG-96, SER-229, and GLU-234; and DE74282670 interacted with LYS-216, SER-229, and HIS-231. These recurring contact features partially overlapped with the interaction profile of the reference inhibitor (FB23), which contacted ARG-96, LYS-216, SER-229, and HIS-231, thereby increasing confidence that these ligands occupied a relevant region of the FTO binding pocket.
Figure 3 Planar interaction diagrams of the 14 selected compounds and FB23 with FTO.
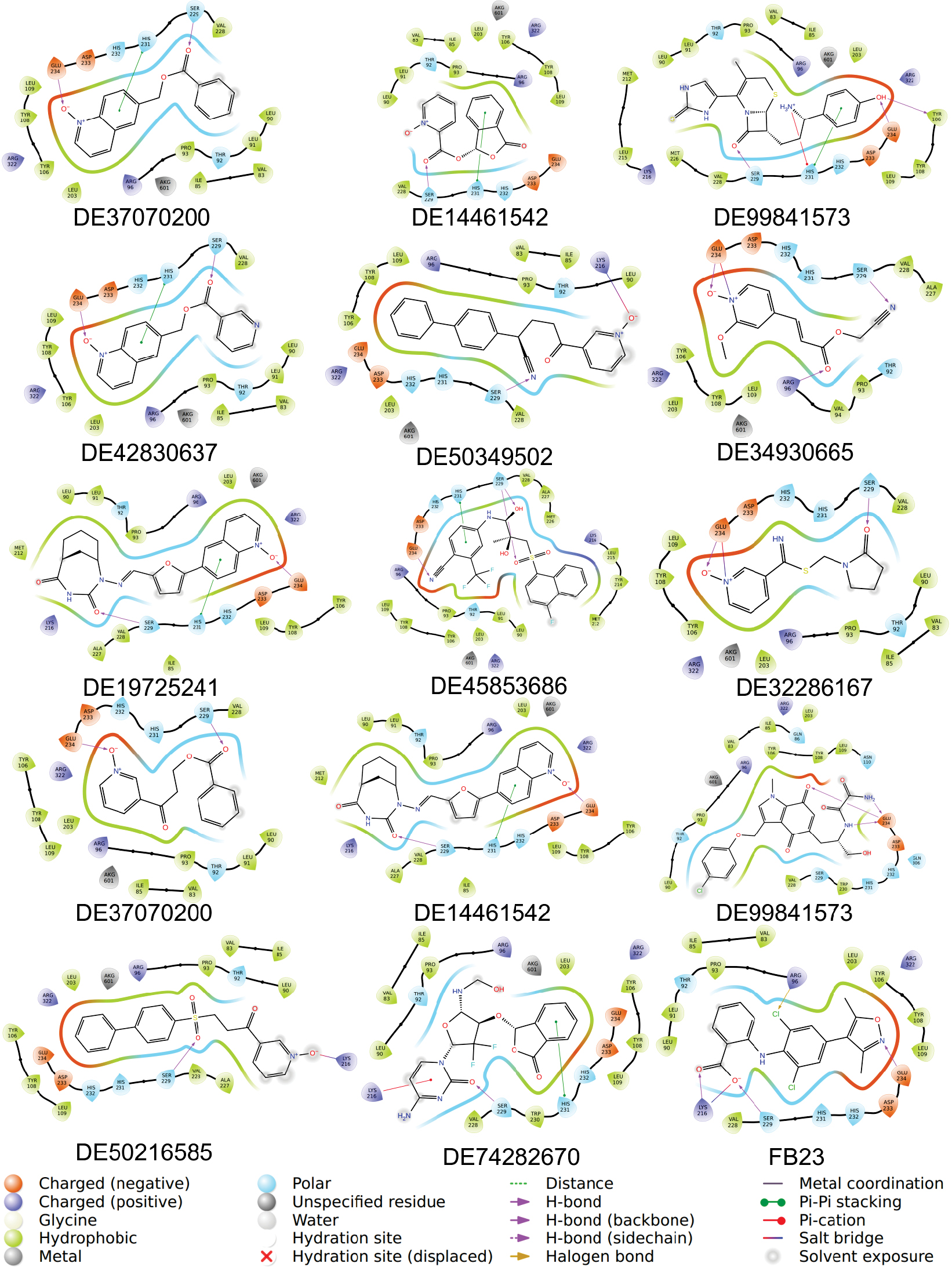
In contrast, DE50349502 and DE50216585 displayed only the limited LYS-216/SER-229 interaction pattern and did not reproduce the broader SER-229-centered interaction network involving HIS-231, GLU-234, or ARG-96 that was observed among the more promising candidates in Figure 3. These restricted contact features suggested weaker anchoring within the binding site and provided less structural support for further prioritization. Therefore, these two compounds were excluded from downstream analysis. DE13072885, despite showing only a GLU-234 contact in the 2D interaction map, was retained because of the highly favorable MMGBSA value, allowing DE13072885to be assessed further alongside the other shortlisted compounds in subsequent BPMD and MD simulations. Taken together, the down-selection was guided not only by docking-related scores but also by the recurrence and coherence of residue-contact patterns revealed by the Figure 3 interaction diagrams.
Evaluating fat mass and obesity-associated protein inhibitor stability through BPMD and conventional MD analyses
A comprehensive analysis of the binding pose metadynamics (BPMD) data, as illustrated in Figure 4A, B, identified DE13072885, DE19725241, and DE74282670 as the highest-priority candidates for subsequent MD simulations based on the combined PoseScore, PersScore, and CompScore profiles. To further assess the robustness of these results, the three prioritized ligands were examined in two independent short MD datasets generated under the same simulation protocol. Across both datasets, DE19725241 consistently showed the lowest ligand RMSD values and the smallest temporal fluctuations, whereas DE74282670 displayed intermediate stability and DE13072885 exhibited the largest deviations. Preservation of this qualitative stability ranking across the two independent datasets supports the robustness of the MD observations.
Figure 4 Verification of compound binding stability over short-term simulations. Dynamic changes in the CV RMSD over time for 13 compounds (A) and bar graphs of 3 different scores (B). The 3 compounds identified by BPMD, the ligand fit on the protein RMSD (C), and the dynamic changes in the ligand RMSD over time (D).
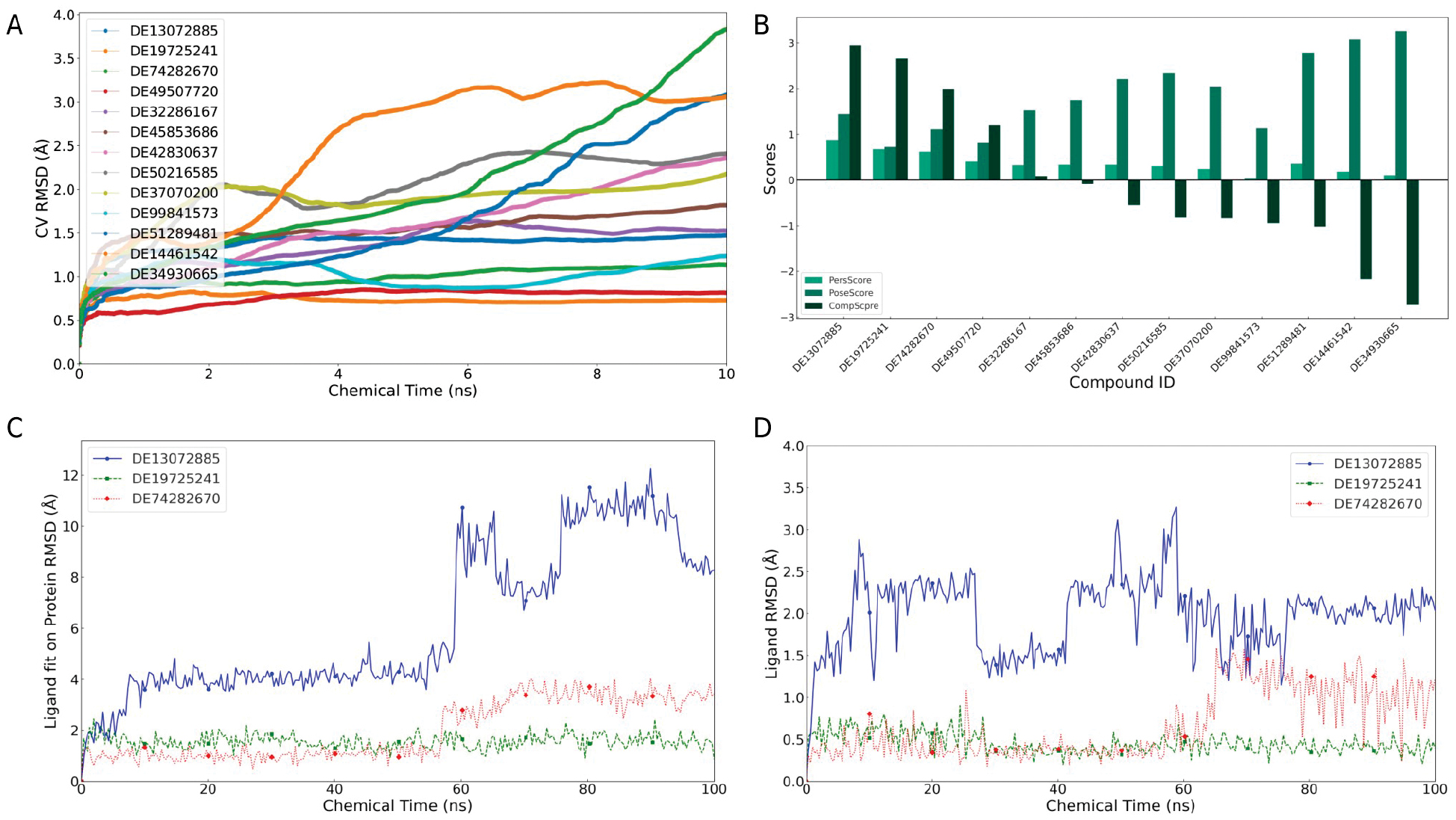
In addition to RMSD, binding stability was also evaluated by the persistence of protein-ligand contacts over the trajectories. DE19725241 maintained more continuous interactions with key FTO pocket residues than the other prioritized compounds, suggesting that the binding mode was not only geometrically stable but also interactionally sustained during the simulations. Taken together, the BPMD scores, replicate consistency in short MD, and persistent contact behavior collectively supported DE19725241 as the most stable candidate for further analysis.
The RMSD was monitored over the course of MD simulations to further evaluate the dynamic stability of these compounds, as shown in Figure 4C, D. The RMSD, which quantifies the temporal deviation of a ligand from the initial bound conformation, serves as a direct indicator of the ligand’s stability within the binding pocket. Compounds that maintain low and stable RMSD values are generally considered to exhibit strong and persistent binding.
DE19725241 consistently demonstrated the most stable binding profile across two independent simulation datasets, which was characterized by the lowest RMSD values and minimal fluctuations over time. This suggests that the compound maintained a well-defined and persistent binding conformation within the FTO pocket. DE74282670 also showed relatively stable binding, although the conformational stability was somewhat lower than DE19725241. In contrast, DE13072885 exhibited higher RMSD values and more pronounced fluctuations in both datasets, particularly in the initial analysis, implying a tendency toward less stable binding and greater deviation from its initial docking pose.
Comparative dynamics and stability analysis of DE19725241 and the reference compound, FB23
The temporal fluctuations in ligand RMSD values was first examined in this in-depth analysis of the dynamic behavior of FB23 and DE19725241 during their interactions with the FTO protein, as illustrated in Figure 5A. The ligand fit the RMSD relative to the FTO protein and the intrinsic RMSD exhibited periodic fluctuations over time for FB23, suggesting a progressive loss of binding site stability. In contrast, DE19725241 displayed a notably stable profile with consistently lower fit and intrinsic RMSD values than FB23, indicating enhanced conformational stability and minimal positional deviation.
Figure 5 Comparison of long-term molecular dynamics simulations of the differences in stability and interactions between DE19725241 and FB23 with FTO. The dynamic changes in RMSD over time for DE19725241 and FB23 (A). Bar graphs showing the interaction statistics of FB23 (B) and DE19725241 (C) with FTO. 2D models of the structure‒activity relationship of FB23 (D) and DE19725241 (E) with FTO.
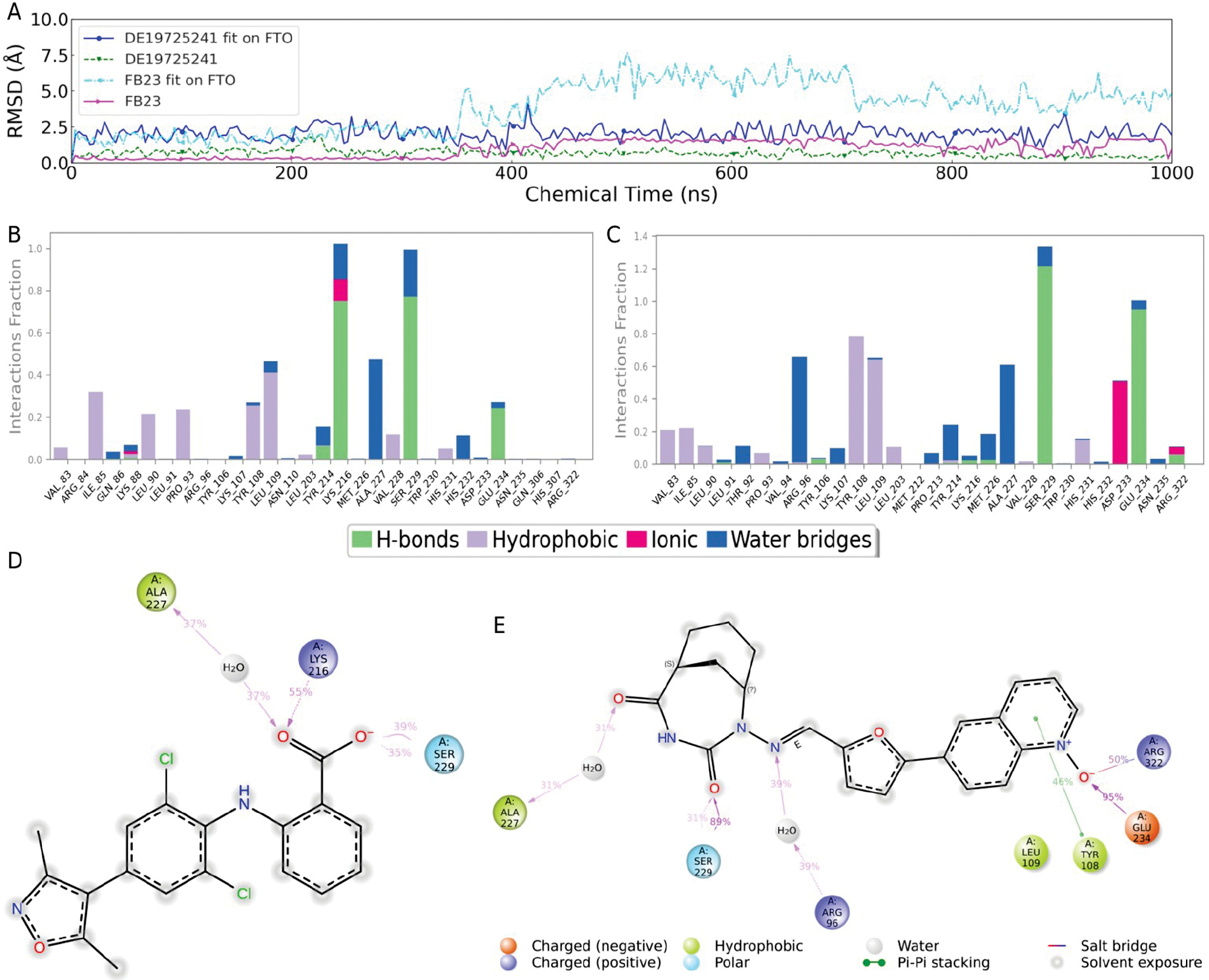
Further examination of the RMSD trends confirmed that DE19725241 maintained consistently lower RMSD fluctuations throughout the simulation period, reflecting the stable accommodation within the FTO binding pocket. This persistent stability underscores the strong conformational compatibility of the ligand with the protein and fidelity to the initial binding pose. Collectively, these results indicated that DE19725241 outperforms FB23 in ligand stability and interaction robustness with FTO. While principal component analysis (PCA) and dynamic cross-correlation maps (DCCMs) can provide a complementary view of collective protein motions, the present work focused on binding-stability descriptors that are directly linked to pocket engagement (ligand RMSD, contact occupancy, and MMGBSA), which are shown in the main figures.
Subsequent analyses of interaction frequencies derived from the simulation trajectories, shown in Figure 5B, C, revealed that DE19725241 formed more frequent interactions with the FTO binding site than FB23. In addition to the RMSD, protein-ligand contact occupancy provided an additional measure of stability. As shown in Figure 5B, C, DE19725241 maintained more persistent contacts with key residues than FB23, particularly ARG-96, GLU-234, and ARG-322, further supporting the robustness of the binding mode throughout the trajectory. Notably, DE19725241 predominantly interacted with ARG-96, GLU-234, and ARG-322, three of the four key substrate-binding residues, suggesting that DE19725241 may exert greater competitive inhibition against the substrate compared to FB23.
Structure–activity relationship (SAR) models were constructed based on residues that maintained interaction frequencies > 30% to further explore the structural basis of binding, as presented in Figure 5D, E. Binding was primarily mediated through its terminal benzoic acid moiety for FB23, which interacted with ALA-227, LYS-216, and SER-229, a pattern consistent with previously reported data, thereby validating the accuracy of our simulation-based SAR model. In contrast, DE19725241 exhibited interactions spanning a broader region of its molecular framework, forming stable contacts with ALA-227, SER-229, ARG-96, TYR-108, GLU-234, and ARG-322, collectively highlighting the superior binding characteristics relative to FB23.
Validating residue significance in DE19725241-fat mass and obesity-associated protein binding through glycine mutation analysis
In silico glycine scanning at key residues implicated in receptor-ligand interactions and repeated MD simulations under consistent conditions were performed to validate the previously established structure-activity relationship (SAR). Although alanine scanning is more common in experimental mutagenesis, glycine was used to enable a uniform minimal side-chain deletion, and importantly, to interrogate ALA-227, which is alanine in the wild type and would be unaffected by alanine substitution. Because glycine can increase local backbone flexibility, the temporal evolution of the protein secondary structure during each mutant simulation was monitored. As shown in Supplementary Figure 1, the overall secondary-structure profile remained stable across all glycine mutants, indicating no global conformational disruption.
Further analyses focusing on ligand fit RMSD values and MMGBSA binding free energies, as illustrated in Figure 6, revealed that mutations at R96G, Y108G, A227G, and R322G substantially increased the RMSD values of DE19725241. This elevation in RMSD indicates a pronounced reduction in binding stability, highlighting the critical roles these residues play in maintaining a stable interaction between DE19725241 and the FTO binding pocket. In contrast, mutations at S229G and E234G led to reduced RMSD values, suggesting an apparent increase in binding stability. However, despite this nominal improvement, the findings suggested that these residues may not be essential for stabilizing the ligand because the increase in RMSD stability did not reflect the functional significance in the native binding context.
Figure 6 Analysis of the stability and binding capacity before and after mutation. Dynamic changes in ligand fit on protein RMSD over time for the wild type and different mutants during the simulation process (A). Dynamic changes in MMGBSA over time for the wild type and different mutants during the simulation process (B).
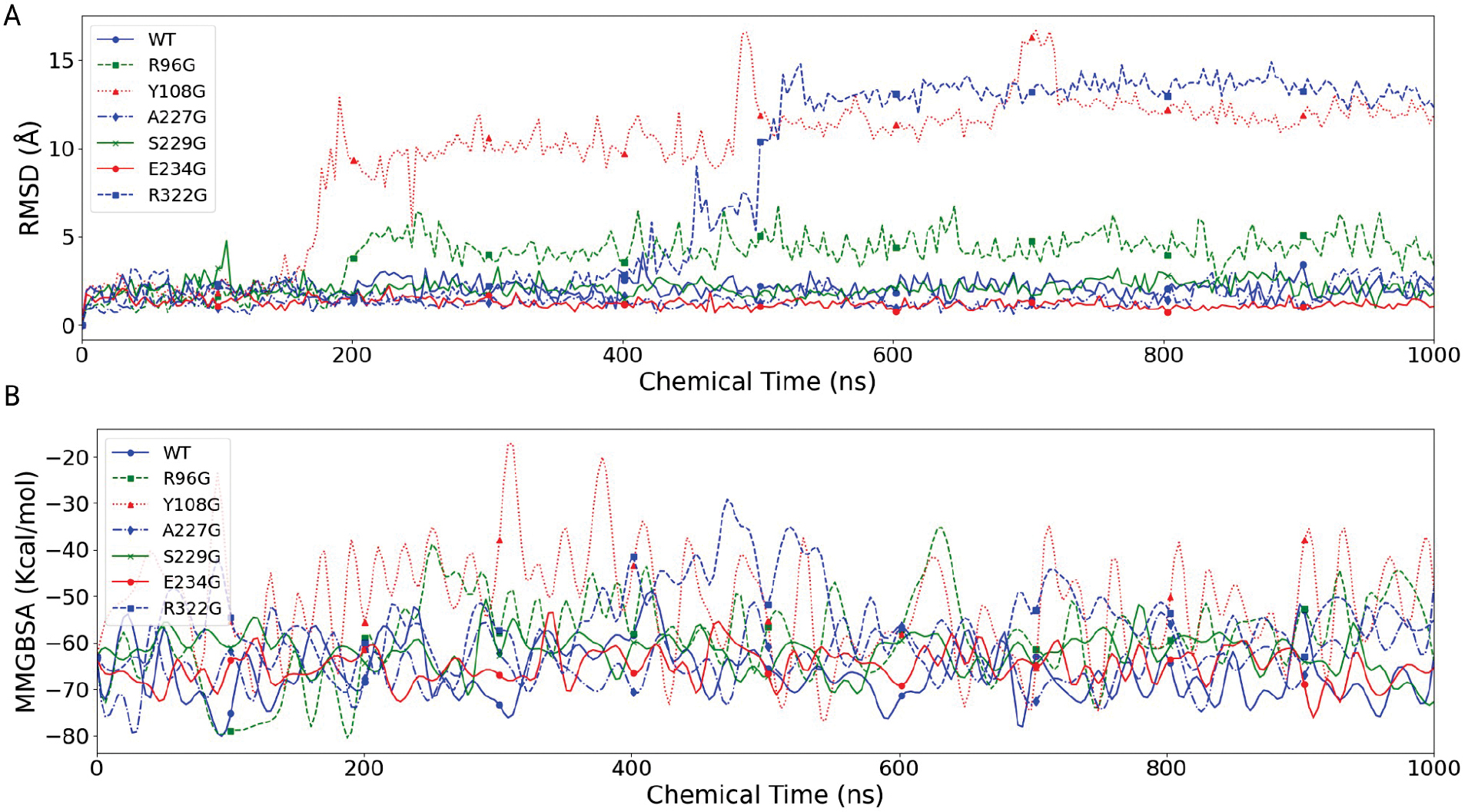
Complementary MMGBSA free energy calculations further supported these observations. Mutations at Y108G, A227G, and R322G resulted in markedly less favorable binding free energies, indicative of weakened ligand affinity and confirming the importance of these residues in mediating high-affinity interactions. Conversely, the S229G mutation was associated with more favorable binding free energy, indicating an increase in binding affinity. Nonetheless, this enhancement suggests that S229 may not be a critical determinant of DE19725241 binding stability under native conditions.
Complementing these observations, the post-mutation interaction models depicted in Figure 7 showed a conspicuous loss of key contacts at the R96G, Y108G, A227G, and R322G sites compared to the wild-type counterparts. This decline in interaction frequency reflects a substantial loss of binding strength and orientation stability, further emphasizing the critical contribution of these residues to ligand anchoring and functional alignment within the FTO active site. Overall, these results highlight that glycine mutations at specific residues severely disrupt the interaction framework necessary for the optimal binding and efficacy of DE19725241, reinforcing the mechanistic insights derived from earlier simulations.
Figure 7 Interaction models of six different mutants with DE19725241. R96G (A), Y108G (B), A227G (C), S229G (D), E234G (E), R322G (F).
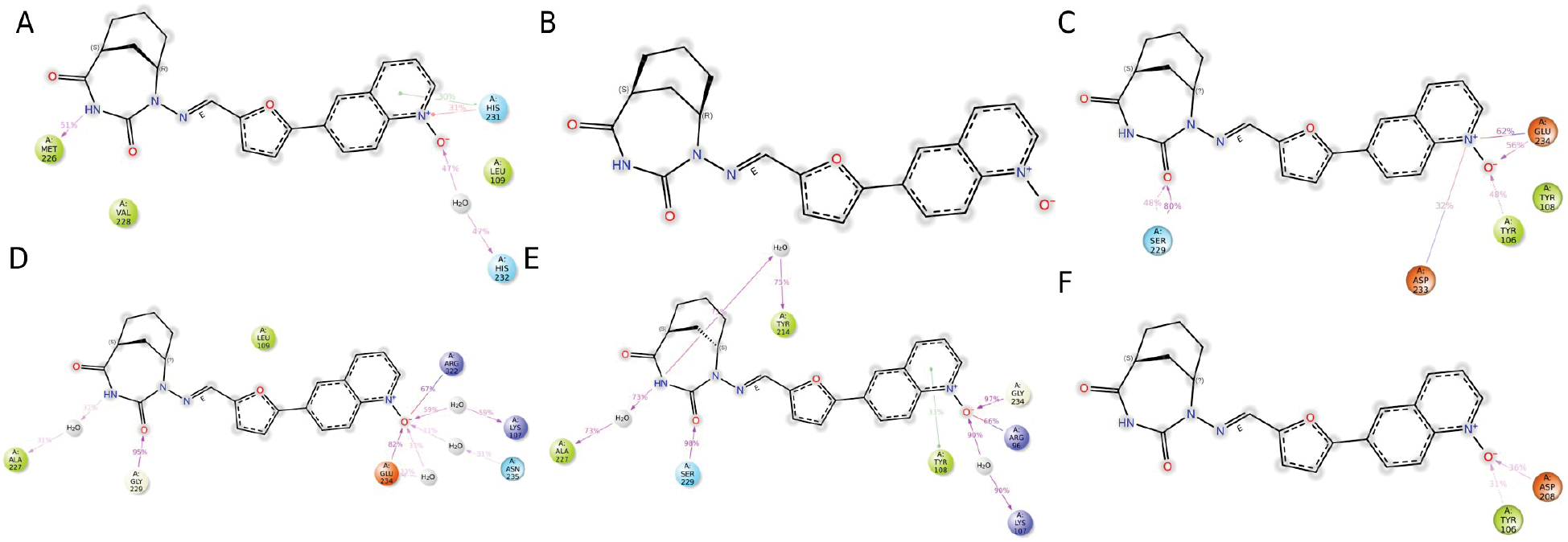
In summary, the results of the integrated analysis confirmed that disrupting R96G, Y108G, A227G, and R322G sites decreased the binding stability and affinity of DE19725241 with the FTO protein, highlighting the importance in preserving the binding strength and stability of DE19725241. Although modifications at the S229G and E234G sites influenced binding stability and affinity, these alterations had minimal effects, suggesting the non-critical role in the original binding profile of DE19725241.
Cross-environmental validation of DE19725241 binding stability in outside-cell, intracellular, and in vivo-like systemic simulations
Whether DE19725241 could maintain a stable binding state with FTO under different simulated environments was further examined after the structure-activity relationship analysis, i.e., outside-cell (OUC), intracellular (INC), and in vivo-like systemic (INV) conditions. To this end, MD simulations were performed in all three environments, followed by analysis of ligand properties and torsional behavior.
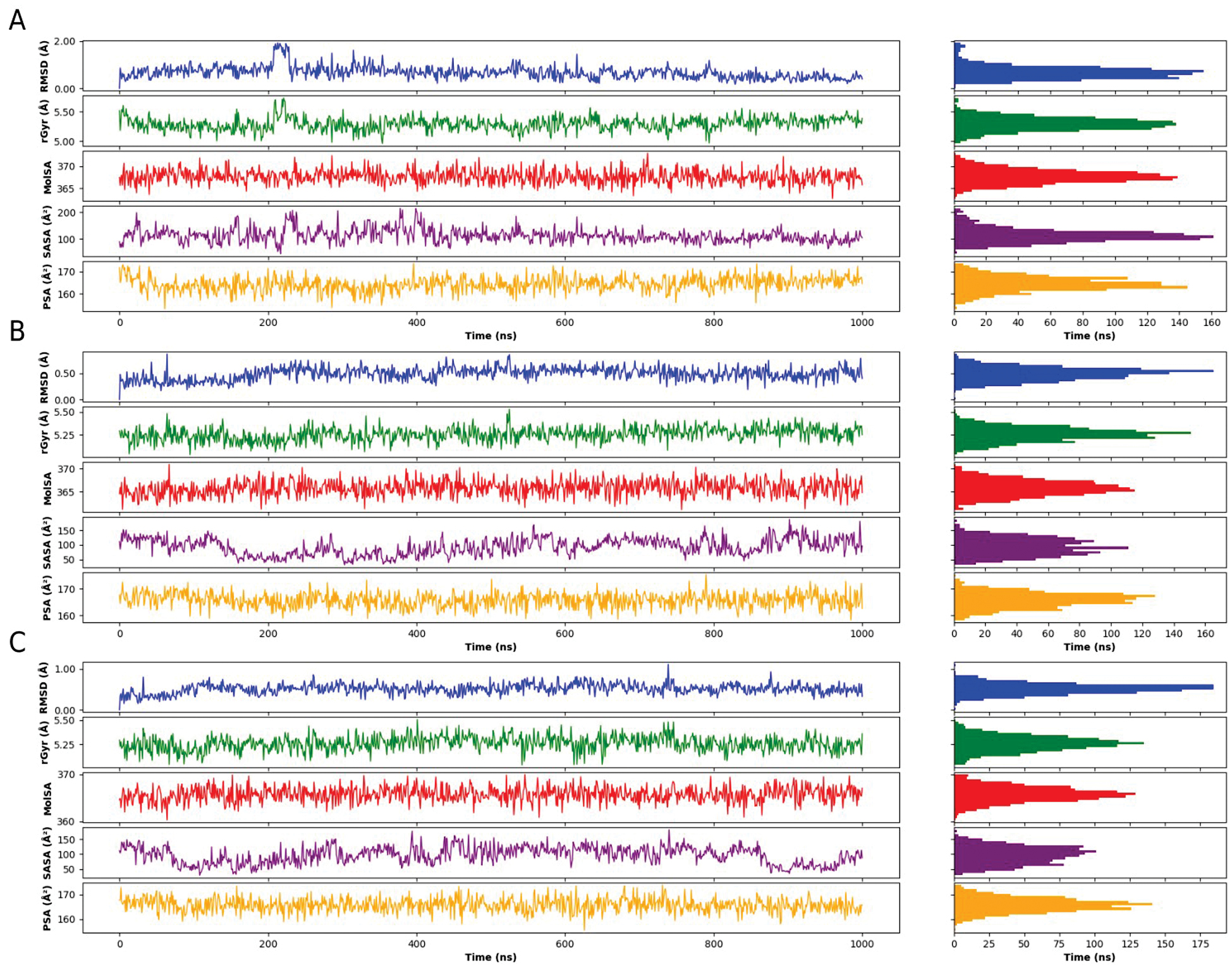
The overall ligand-property profiles of DE19725241 remained comparable across the OUC, INC, and INV simulations, as shown in Figure 8. In particular, the RMSD fluctuations of the ligand were narrower in the INC and INV systems than the OUC system, suggesting that the bound conformation of DE19725241 remained stable under the more physiologically relevant conditions. At the same time, other descriptors, including the radius of gyration (rGyr), molecular surface area (MolSA), solvent-accessible surface area (SASA), and polar surface area (PSA), showed no major shifts among the three environments. These results indicated that DE19725241 preserved overall compactness and surface-exposure characteristics after binding to FTO, despite environmental changes.
Figure 8 Ligand properties of DE19725241 after binding with FTO in three different environments. OUC (A), INC (B), INV (C).
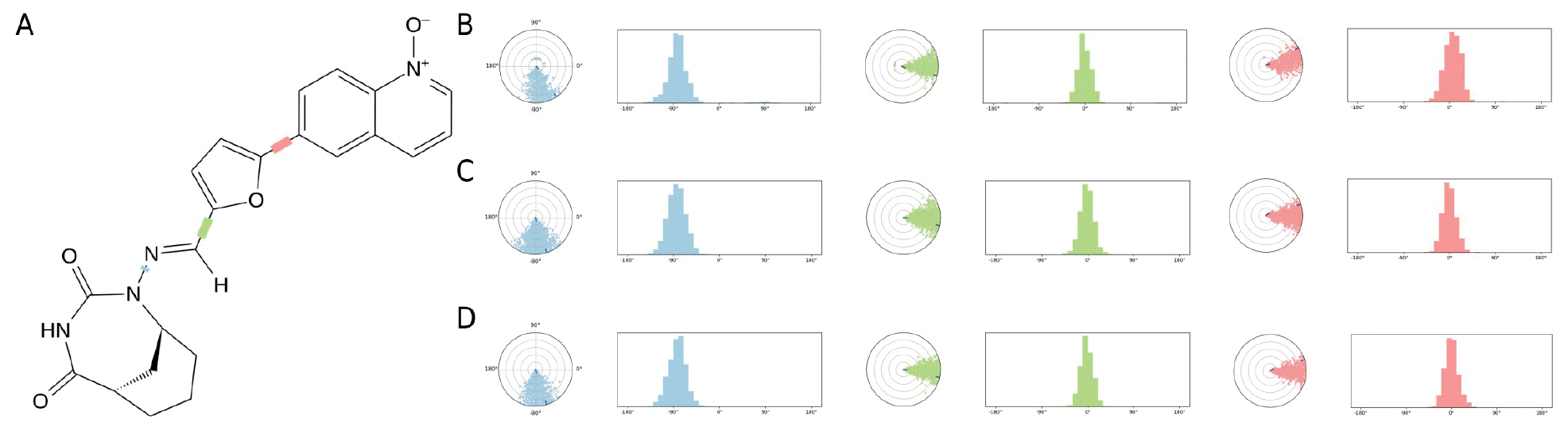
The torsional analysis further supported this conclusion. DE19725241 maintained a defined overall distribution of torsion keys, as illustrated in Figure 9A-D show that the three selected torsion angles remained within comparable ranges in the OUC, INC, and INV simulations. This result suggests that the ligand retains moderate conformational flexibility while avoiding large torsional deviations in different environments. Taken together, the ligand-property analysis in Figure 8 and the torsion-key analysis in Figure 9 suggest that DE19725241 can maintain a stable and adaptable binding conformation with FTO across multiple simulated biological environments.
Figure 9 Analysis of torsion keys of DE19725241 after binding with FTO in three different environments. The overall distribution of torsion keys for DE19725241 (A). The range of torsion angles for three torsion keys of DE19725241 in the OUC environment (B), the INC environment (C), and the INV environment (D).
Comprehensive ADMET profile analysis for evaluating the therapeutic potential of DE19725241
ADMET profiling provides an initial estimate of the absorption, distribution, metabolism, excretion, and toxicity properties that may influence the developability of a lead compound. To facilitate interpretation of the full prediction output, the representative ADMET endpoints of DE19725241 are summarized in Table 3, while the complete results are provided in Supplementary Table 1.
Table 3 Representative Predicted ADMET Properties of DE19725241
| Category | Endpoint | Predicted Profile | Main Implication |
|---|---|---|---|
| Absorption | PAMPA permeability | High | Favorable passive membrane diffusion |
| Absorption | Caco-2 permeability | Low | Potential intestinal absorption liability |
| Transporter | P-gp substrate/inhibitor | No/No | P-gp is unlikely to explain the low Caco-2 signal |
| Distribution | Plasma protein binding | 61.90% | Moderate free fraction in circulation |
| Distribution | Volume of distribution | 0.553 L/kg | Limited tissue distribution |
| Distribution | BBB penetration | Low | Low predicted CNS exposure |
| Metabolism | CYP3A4 substrate | Yes | Likely metabolic liability |
| Metabolism | Human liver microsome stability | Low | Rapid hepatic turnover predicted |
| Metabolism | CYP inhibition | CYP1A2 high; CYP2B6/CYP2C8 weaker | Possible drug-drug interaction risk |
| Excretion | Clearance | 4.042 L/h | Rapid systemic clearance |
| Excretion | Half-life | 0.848 h | Short predicted exposure window |
| Toxicity | Genotoxicity/carcinogenicity/mutagenicity | Low risk | Generally favorable toxicity profile |
| Toxicity | Skin sensitization | Potential concern | Liability requiring follow-up optimization |
| Environment | Biodegradability | Non-biodegradable | Environmental liability |
Overall, DE19725241 combines several favorable features with clear developability liabilities. The compound showed high PAMPA permeability, suggesting favorable passive diffusion but low Caco-2 permeability, indicating a potential limitation in intestinal uptake under more physiologically relevant conditions. Because DE19725241 was predicted to be neither a substrate nor an inhibitor of P-glycoprotein, this discrepancy may reflect the influence of other transport processes or intracellular metabolism. Plasma protein binding was moderate (61.9%) in distribution-related predictions, whereas the low predicted volume of distribution (0.553 L/kg) suggested limited tissue penetration and low blood-brain barrier exposure.
The predicted metabolic profile indicated that DE19725241 is primarily a CYP3A4 substrate and has low human liver microsome stability, which is consistent with rapid hepatic turnover. In addition, the predicted inhibition of CYP1A2 with weaker effects on CYP2B6 and CYP2C8, suggested a possible risk of drug-drug interactions. These features were aligned with the predicted excretion profile, which showed relatively rapid clearance (4.042 L/h) and a short half-life (0.848 h). Toxicity-related predictions were generally favorable with low predicted risks of genotoxicity, carcinogenicity, and mutagenicity, although potential skin sensitization and non-biodegradability remained concerns.
From a computation-only perspective, the most immediate next steps are to perform metabolism-site prediction and analog enumeration to reduce CYP3A4 liability, apply multi-parameter optimization to balance binding with Caco-2 permeability and microsomal stability, and run exposure-oriented PK/PBPK simulations to assess whether the predicted clearance and half-life can be improved before experimental follow-up.
CCK-8-based evaluation of the anti-proliferative activity of DE19725241 in pancreatic cancer cells
CCK-8 assays were performed on three pancreatic cancer cell lines (MIA PaCa-2, BxPC-3, and PANC-1) and a normal pancreatic ductal epithelial cell line (hTERT-HPNE) across a concentration range of 0-100 μM to evaluate the effects of DE19725241 on pancreatic cancer cell viability, as shown in Figure 10. Each condition was tested in six biological replicates, and cell viability was expressed as a percentage relative to the untreated control.
Figure 10 CCK-8-based dose-response curves of DE19725241 in pancreatic cancer and normal pancreatic epithelial cell lines. (A) MiaPaCa-2 cells; (B) BxPC-3 cells; (C) PANC-1 cells; (D) hTERT-HPNE cells. Cells were treated with the indicated concentrations of DE19725241 for 48 h and cell viability was measured using the CCK-8 assay. Data are presented as the mean ± SD from six replicates. Dose-response curves were generated by four-parameter nonlinear regression.
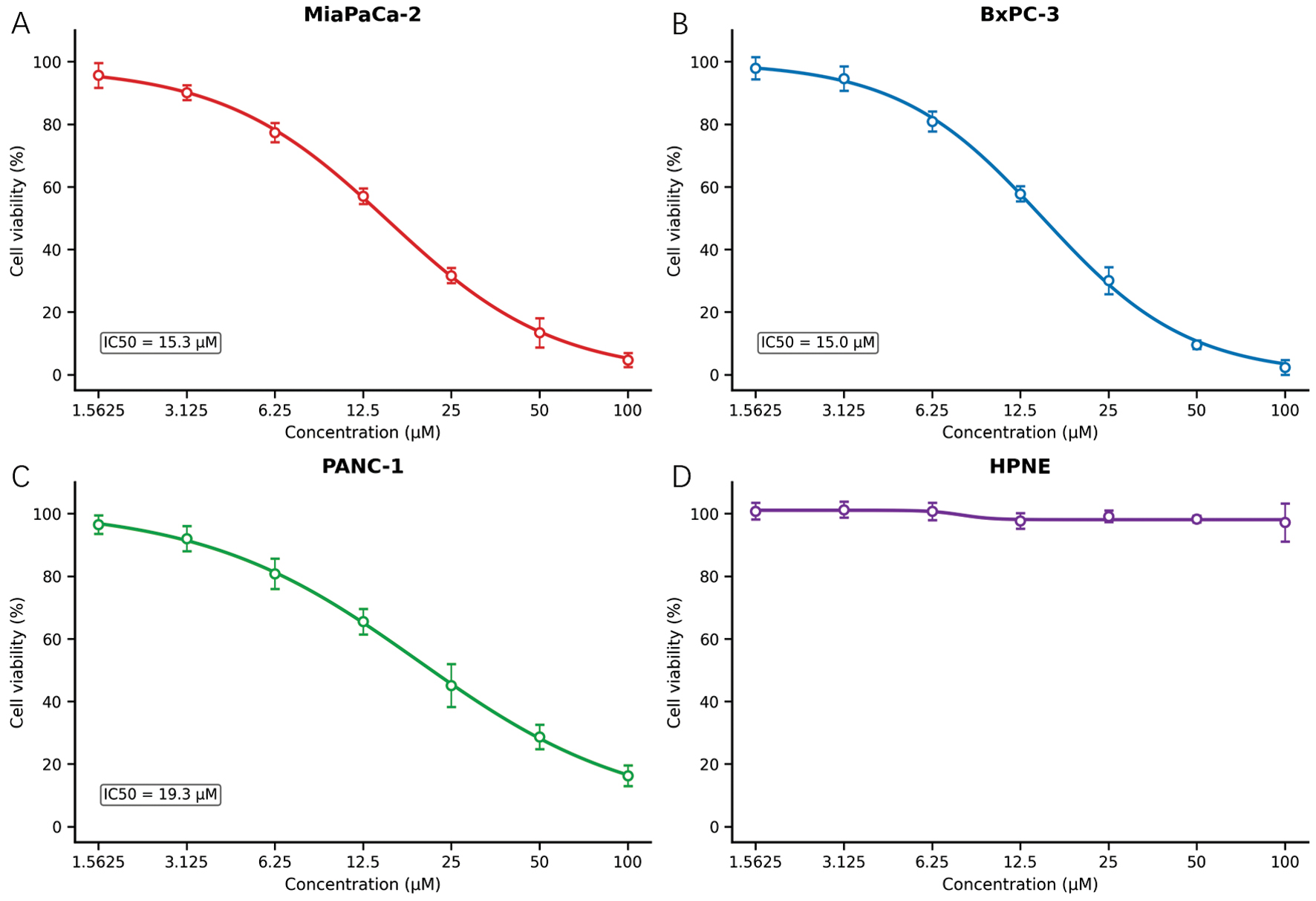
The compound demonstrated dose-dependent growth inhibition in all three pancreatic cancer cell lines, whereas hTERT-HPNE cells showed minimal sensitivity. Non-linear regression using a four-parameter logistic model was used to estimate IC50 values. Among the tested cancer cell lines, MiaPaCa-2 and BxPC-3 exhibited IC50 values of approximately 15 μM, whereas PANC-1 showed an IC50 of approximately 20 μM, indicating cell line-specific variation in susceptibility. In contrast, hTERT-HPNE cells showed only approximately 5% growth inhibition, even at 100 μM, suggesting a favorable in vitro selectivity window.
These results collectively support moderate but selective anti-proliferative activity of DE19725241 against pancreatic cancer cells, while largely sparing normal pancreatic epithelium and providing a quantitative basis for subsequent mechanistic and in vivo investigations.
Discussion
FTO [43], an enzyme that functions as m6A RNA demethylase, has garnered significant attention because of its role in oncogenic processes across several cancers, including pancreatic cancer [44], breast cancer [45], and AML [23]. Moreover, FTO is recognized as an important regulatory factor that governs cancer stem cell maintenance and immune evasion mechanisms [46]. In the present study DE19725241 was identified as a candidate FTO-targeting compound through a computer-aided discovery workflow that integrated transcriptomic target prioritization, large-scale virtual screening, molecular simulations, and cell-based phenotypic evaluation. This section contextualizes DE19725241 within the broader landscape of FTO-focused drug discovery and discusses the limitations that must be addressed before translational development can be considered.
Recent advances in the understanding of the FTO biological functions highlight the involvement in modulating epigenetic landscapes and influencing cellular metabolism, survival, and proliferation. FTO dysregulation has been implicated in the pathogenesis of several cancers, making FTO a promising target for therapeutic intervention [46]. Prior studies have identified FTO inhibitors, such as FB23 and FB23-2, that can modulate m6A levels, thereby affecting mRNA stability and gene expression [30]. However, these molecules have shown limited clinical utility because of the suboptimal pharmacokinetic profiles and insufficient therapeutic efficacy [47].
Therefore, DE19725241 should be regarded as a promising candidate for further investigation rather than a definitively validated FTO inhibitor. The compound showed favorable predicted affinity for the FTO catalytic pocket and maintained stable interactions with key residues in molecular simulations. In addition, glycine mutation analysis supported the contribution of several critical residues to the proposed binding mode. Although DE19725241 showed more favorable predicted interactions than FB23 in our simulations, these findings should be interpreted as computational prioritization rather than proof of superior cellular potency relative to optimized benchmark inhibitors.
The pharmacokinetic and pharmacodynamic properties of DE19725241 suggest both encouraging features and substantial translational liabilities compared to earlier FTO-directed compounds. Initial ADMET assessments indicated acceptable passive permeability and a generally manageable predicted toxicity profile but also highlight several pharmacokinetic concerns that limit the immediate therapeutic viability of the current scaffold. In particular, DE19725241 is predicted to be primarily metabolized by CYP3A4, to have low stability in human liver microsomes, to inhibit CYP1A2 and to a lesser extent CYP2B6 and CYP2C8, and to undergo rapid systemic elimination with a predicted plasma clearance of 4.042 L/h and a short half-life of 0.848 h. Together, these features raise concerns about insufficient drug exposure, the need for frequent dosing, and possible drug-drug interactions. Accordingly, although DE19725241 remains a promising scaffold for further study, DE19725241 should be regarded as a moderate-activity lead rather than an optimized inhibitor or a clinically ready compound.
These pharmacologic considerations are especially important for pancreatic cancer, where systemic therapies often operate within narrow therapeutic windows. Taken together, the current computational, ADMET, and cell-based data indicate that DE19725241 should be viewed as an early-stage hit/lead scaffold, the predicted binding behavior and moderate anti-proliferative activity of which justify further optimization, rather than as a compound already positioned for therapeutic application.
If future studies confirm on-target FTO modulation, the potential of DE19725241 to participate in combination therapy strategies would merit further consideration. Given the complexity of cancer biology, which is characterized by redundant and adaptive oncogenic pathways, multimodal therapeutic approaches are often necessary. For example, the use of trastuzumab in combination with chemotherapy for HER2-positive breast cancer dramatically improves patient outcomes by targeting both the HER2 receptor and cancer cell division [48]. Similarly, combining DE19725241 with other anticancer agents, particularly those that target complementary molecular pathways or modulate the immune microenvironment, could become a reasonable strategy for future evaluation.
One notable example of an effective combination therapy is the combination of nivolumab, a PD-1 immune checkpoint inhibitor, with ipilimumab, a CTLA-4 inhibitor, in melanoma treatment. This strategy leverages dual immune modulation to more effectively combat tumor cells and has successfully improved patient survival rates [49]. By targeting different aspects of the immune response, this strategy helps overcome the tumor-induced immunosuppression that often limits the effectiveness of single-agent immunotherapies.
Similarly, if DE19725241 is confirmed to modulate FTO-dependent signaling, integrating DE19725241 with existing therapies that disrupt different signaling pathways or enhance immune responses could provide a rational way to explore resistance-prevention strategies. For example, combining an FTO-targeting compound with agents that target the PI3K/AKT pathway, which is activated in many cancers, including pancreatic cancer, might provide a synergistic effect if FTO-dependent metabolic regulation is confirmed experimentally.
Moreover, considering the role of FTO in regulating the immune microenvironment by modulating m6A levels on mRNA transcripts encoding immune checkpoints, DE19725241 could potentially be evaluated in combination with immune checkpoint inhibitors once the on-target mechanism is clarified. The modulation of m6A levels influences the expression of various immune-related genes, potentially affecting the efficacy of immunotherapies.
While the identification of DE19725241 as a computationally and phenotypically supported FTO-targeting candidate is encouraging, several major limitations must be acknowledged. Most importantly, the current study does not provide direct biochemical or cellular evidence demonstrating that DE19725241 inhibits FTO demethylase activity. Exploratory follow-up m6A-seq analyses after compound treatment suggested bidirectional methylation changes but these observations were not orthogonally validated and therefore were not interpreted as definitive target-engagement evidence. In addition, although CCK-8 assays yielded IC50 values of approximately 15-22 μM in pancreatic cancer cell lines, this level of activity remains moderate by conventional drug-discovery standards. Preliminary attempts to establish a purified-protein inhibition assay also did not yield sufficiently robust and reproducible data for mechanistic interpretation.
Another major limitation of the present study is the lack of in vivo validation. Although DE19725241 showed favorable computational binding features and selective anti-proliferative activity in pancreatic cancer cell lines, the antitumor efficacy, pharmacokinetic behavior, tolerability, and systemic safety remain unknown in whole-animal settings. Therefore, the current findings should be interpreted as an early-stage discovery rather than preclinical therapeutic validation. Future studies should evaluate DE19725241 in PDAC xenograft or other relevant in vivo models to determine whether the observed in vitro and in silico properties translate into meaningful antitumor activity in vivo.
In addition, although DE19725241 showed selective anti-proliferative activity in pancreatic cancer cells relative to HPNE cells, this selectivity alone does not exclude off-target mechanisms. Future studies should therefore incorporate genetic rescue approaches, such as FTO overexpression or knockdown, together with broader counter-screen profiling and direct target-engagement assays to determine whether the observed cellular phenotype is specifically mediated through FTO inhibition.
Another important consideration is the risk of drug-drug interactions, particularly in the context of combination therapies, which are often required to overcome resistance mechanisms in oncology. This issue is especially relevant for DE19725241 because the predicted inhibition of CYP1A2, together with weaker effects on CYP2B6 and CYP2C8, could alter the pharmacokinetics of co-administered agents. Even though DE19725241 exhibits encouraging computational and phenotypic properties, the interaction with other therapeutic agents, especially those affecting related metabolic pathways, must be carefully evaluated in future studies.
Finally, the predicted rapid plasma clearance and short half-life suggest that maintaining effective systemic exposure may be difficult without further optimization. To overcome these hurdles, both medicinal chemistry and formulation-based strategies should be considered. On the chemistry side, scaffold-focused optimization aimed at improving microsomal stability, reducing CYP inhibition, and modifying metabolically vulnerable moieties may help extend exposure and reduce interaction risk. On the formulation side, sustained-release systems, nanoparticle or liposomal delivery approaches, and alternative routes of administration could be explored if conventional dosing proves insufficient to maintain therapeutic concentrations. Until such work is completed, the translational potential of DE19725241 should be interpreted cautiously.
Extensive preclinical studies should be conducted to validate the efficacy and safety profile of DE19725241 across a variety of cancer models in the future. These studies should aim to elucidate the on-target mechanism of action of the compound through direct enzymatic inhibition assays, m6A dot blot or LC-MS/MS-based global m6A quantification, targeted validation of established FTO-regulated transcripts, and genetic rescue approaches, such as FTO overexpression or knockdown. In parallel, scaffold-focused medicinal chemistry optimization will likely be necessary to improve potency, reduce CYP-related liabilities, and evaluate possible metabolic instability of the current chemotype. Dedicated in vivo pharmacokinetic studies and exploratory formulation development will also be important to determine whether systemic exposure can be improved sufficiently for therapeutic application.
In conclusion, the identification of DE19725241 as a candidate FTO-targeting compound represents a meaningful step forward in transcriptome-guided drug discovery. By integrating large-scale computational screening with transcriptomic analysis and in vitro phenotypic validation, this study provides a framework for prioritizing epitranscriptomic regulators implicated in cancer progression and immune modulation. However, DE19725241 is currently an early-stage lead scaffold rather than a clinically viable inhibitor given the lack of direct target-engagement evidence, the absence of in vivo validation, and the predicted pharmacokinetic liabilities related to metabolism, clearance, and half-life. Direct biochemical and cellular confirmation of the on-target mechanism, together with medicinal chemistry refinement, pharmacokinetic optimization, and further translational studies, will be essential before clinical advancement.
Conclusion
In this study DE19725241 was identified as a candidate FTO-targeting compound relevant to pancreatic cancer through an integrative approach combining transcriptomic analysis, active learning-assisted virtual screening, and molecular dynamics simulations. DE19725241 demonstrated favorable predicted binding affinity and stability compared to reference compounds and showed moderate, selective anti-proliferative activity in pancreatic cancer cell lines. However, substantial challenges remain, including the need for direct mechanistic validation, in vivo efficacy and safety studies, and optimization of pharmacokinetic liabilities, such as rapid clearance, short half-life, and CYP-related interaction risks. These findings therefore position DE19725241 as an early-stage scaffold for further refinement rather than a therapeutically ready inhibitor, while still underscoring the value of transcriptome-guided, computer-aided drug discovery in precision oncology.
Data availability statement
The datasets generated and/or analyzed during the current study are available from the corresponding author upon reasonable request.
Ethics statement
No direct interactions with human or animal subjects were involved. Therefore, ethical approval and informed consent were not required.
Author contributions
The authors confirm the following contributions to the article. Study conception and design: Chengjie Xu, Chaojie Huang, and Fangyu Ren; data collection: Chengjie Xu, Zhikang Cai, Donglin Wang, and Rui Chen; analysis and interpretation of results: Chengjie Xu; draft manuscript preparation: Chengjie Xu and Fangyu Ren. All authors reviewed the results and approved the final version of the manuscript.
Funding
This work was supported by the Medical Health Science and Technology Project of Zhejiang Provincial Health Commission (Grant No. 2022RC178) and the “Pioneer” and “Leading Goose” R&D Program of Zhejiang Province (Grant No. 2025C2057).
Conflict of interest
The authors declare that there are no conflicts of interest.
Supplementary materials
Supplementary Material can be downloaded from https://bio-integration.org/wp-content/uploads/2026/07/bioi20250225_Supplemental.pdf.
Graphical abstract
Highlights
- FTO is overexpressed in pancreatic cancer and predicts poorer overall survival.
- Active learning prioritized FTO-targeting candidates from a 22.6-million-compound library.
- DE19725241 showed stable predicted binding to key FTO pocket residues across simulations.
- DE19725241 selectively inhibited pancreatic cancer cell growth while sparing normal pancreatic epithelial cells.
- Findings support DE19725241 as an early scaffold requiring mechanistic and in vivo validation.
In brief
Using transcriptomic analysis and active learning-assisted screening of 22.6 million compounds, this study identifies DE19725241 as a candidate FTO-targeting scaffold in pancreatic cancer. The compound showed favorable computational binding behavior and selective, moderate antiproliferative activity, but requires direct target-engagement, pharmacokinetic, and in vivo validation before therapeutic development.
References
- Bugazia D, Al-Najjar E, Esmail A, Abdelrahim S, Abboud K, et al. Pancreatic ductal adenocarcinoma: the latest on diagnosis, molecular profiling, and systemic treatments. Front Oncol 2024;14:1386699. [PMID: 39011469 DOI: 10.3389/fonc.2024.1386699]
- Halbrook CJ, Lyssiotis CA, Pasca di Magliano M, Maitra A. Pancreatic cancer: advances and challenges. Cell 2023;186(8):1729-54. [PMID: 37059070 DOI: 10.1016/j.cell.2023.02.014]
- Mottini C, Auciello FR, Manni I, Pilarsky C, Caputo D, et al. The cross-talk between the macro and micro-environment in precursor lesions of pancreatic cancer leads to new and promising circulating biomarkers. J Exp Clin Cancer Res 2024;43(1):198. [PMID: 39020414 DOI: 10.1186/s13046-024-03117-5]
- Qing T, Mohsen H, Marczyk M, Ye Y, O’Meara T, et al. Germline variant burden in cancer genes correlates with age at diagnosis and somatic mutation burden. Nat Commun 2020;11(1):2438.
- Shin H, Choi BH, Shim O, Kim J, Park Y, et al. Single test-based diagnosis of multiple cancer types using Exosome-SERS-AI for early stage cancers. Nat Commun 2023;14(1):1644. [PMID: 36964142 DOI: 10.1038/s41467-023-37403-1]
- Golivi Y, Kumari S, Farran B, Alam A, Peela S, et al. Small molecular inhibitors: therapeutic strategies for pancreatic cancer. Drug Discov Today 2024;29(7):104053. [PMID: 38849028 DOI: 10.1016/j.drudis.2024.104053]
- Wang Y, Bui TA, Yang X, Hutvagner G, Deng W. Advancements in gene therapies targeting mutant KRAS in cancers. Cancer Metastasis Rev 2025;44(1):24. [PMID: 39820726 DOI: 10.1007/s10555-025-10243-9]
- Sato H, Sasaki K, Hara T, Tsuji Y, Arao Y, et al. Pancreatic cancer research beyond DNA mutations. Biomolecules 2022;12(10):1503. [PMID: 36291712 DOI: 10.3390/biom12101503]
- Jones L, Cunningham D, Starling N. HER-2 directed therapies across gastrointestinal tract cancers – a new frontier. CancerTreat Rev 2024;129:102789. [PMID: 38959629 DOI: 10.1016/j.ctrv.2024.102789]
- Gulwani D, Upadhyay P, Goel R, Sarangthem V, Singh TD. Nanomedicine mediated thyroid cancer diagnosis and treatment: an approach from generalized to personalized medicine. Discov Oncol 2024;15(1):789. [PMID: 39692930 DOI: 10.1007/s12672-024-01677-8]
- Singh S, Sadhukhan S, Sonawane A. 20 years since the approval of first EGFR-TKI, gefitinib: insight and foresight. Biochim Biophys Acta Rev Cancer 2023;1878(6):188967. [PMID: 37657684 DOI: 10.1016/j.bbcan.2023.188967]
- Yang Z, Hackshaw A, Feng Q, Fu X, Zhang Y, et al. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: a meta-analysis. Int J Cancer 2017;140(12):2805-19. [PMID: 28295308 DOI: 10.1002/ijc.30691]
- Waters AM, Der CJ. KRAS: The critical driver and therapeutic target for pancreatic cancer. Cold Spring Harb Perspect Med 2018;8(9):a031435. [PMID: 29229669 DOI: 10.1101/cshperspect.a031435]
- Braal CL, Jongbloed EM, Wilting SM, Mathijssen RHJ, Koolen SLW, et al. Inhibiting CDK4/6 in breast cancer with palbociclib, ribociclib, and abemaciclib: similarities and differences. Drugs 2021;81(3):317-31. [PMID: 33369721 DOI: 10.1007/s40265-020-01461-2]
- Mughal MJ, Bhadresha K, Kwok HF. CDK inhibitors from past to present: a new wave of cancer therapy. Semin Cancer Biol 2023;88:106-22. [PMID: 36565895 DOI: 10.1016/j.semcancer.2022.12.006]
- Mortazavi M, Moosavi F, Martini M, Giovannetti E, Firuzi O. Prospects of targeting PI3K/AKT/mTOR pathway in pancreatic cancer. Crit Rev Oncol Hematol 2022;176:103749. [PMID: 35728737 DOI: 10.1016/j.critrevonc.2022.103749]
- Yu L, Wei J, Liu P. Attacking the PI3K/Akt/mTOR signaling pathway for targeted therapeutic treatment in human cancer. Semin Cancer Biol 2022;85:69-94. [PMID: 34175443 DOI: 10.1016/j.semcancer.2021.06.019]
- Mayuri K, Varalakshmi D, Tharaheswari M, Somala CS, Priya SS, et al. Identifying potent fat mass and obesity-associated protein inhibitors using deep learning-based hybrid procedures. BioMedInformatics 2024;4(1):347-59.
- Frye M, Harada BT, Behm M, He C. RNA modifications modulate gene expression during development. Science 2018;361(6409):1346-9. [PMID: 30262497 DOI: 10.1126/science.aau1646]
- Jia G, Fu Y, Zhao X, Dai Q, Zheng G, et al. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol 2011;7(12):885-7. [PMID: 22002720 DOI: 10.1038/nchembio.687]
- Jia G, Fu Y, He C. Reversible RNA adenosine methylation in biological regulation. Trends Genet 2013;29(2):108-15. [PMID: 23218460 DOI: 10.1016/j.tig.2012.11.003]
- Cao K, Du Y, Bao X, Han M, Su R, et al. Glutathione-bioimprinted nanoparticles targeting of N6-methyladenosine FTO demethylase as a strategy against leukemic stem cells. Small 2022;18(13):e2106558. [PMID: 35119204 DOI: 10.1002/smll.202106558]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, et al. R-2HG exhibits anti-tumor activity by targeting FTO/m6A/MYC/CEBPA signaling. Cell 2018;172(1-2):90-105.e23. [PMID: 29249359 DOI: 10.1016/j.cell.2017.11.031]
- Li Y, Su R, Deng X, Chen Y, Chen J. FTO in cancer: functions, molecular mechanisms, and therapeutic implications. Trends Cancer 2022;8(7):598-614. [PMID: 35346615 DOI: 10.1016/j.trecan.2022.02.010]
- Chen B, Ye F, Yu L, Jia G, Huang X, et al. Development of cell-active N6-methyladenosine RNA demethylase FTO inhibitor. J Am Chem Soc 2012;134(43):17963-71. [PMID: 23045983 DOI: 10.1021/ja3064149]
- He W, Zhou B, Liu W, Zhang M, Shen Z, et al. Identification of a novel small-molecule binding site of the fat mass and obesity associated protein (FTO). J Med Chem 2015;58(18):7341-8. [PMID: 26314339 DOI: 10.1021/acs.jmedchem.5b00702]
- Huang Y, Yan J, Li Q, Li J, Gong S, et al. Meclofenamic acid selectively inhibits FTO demethylation of m6A over ALKBH5. Nucleic Acids Res 2015;43(1):373-84. [PMID: 25452335 DOI: 10.1093/nar/gku1276]
- Singh B, Kinne HE, Milligan RD, Washburn LJ, Olsen M, et al. Important role of FTO in the survival of rare panresistant triple-negative inflammatory breast cancer cells facing a severe metabolic challenge. PLoS One 2016;11(7):e0159072. [PMID: 27390851 DOI: 10.1371/journal.pone.0159072]
- Wang T, Hong T, Huang Y, Su H, Wu F, et al. Fluorescein derivatives as bifunctional molecules for the simultaneous inhibiting and labeling of FTO protein. J Am Chem Soc 2015;137(43):13736-9. [PMID: 26457839 DOI: 10.1021/jacs.5b06690]
- Huang Y, Su R, Sheng Y, Dong L, Dong Z, et al. Small-molecule targeting of oncogenic FTO demethylase in acute myeloid leukemia. Cancer Cell 2019;35(4):677-91.e10. [PMID: 30991027 DOI: 10.1016/j.ccell.2019.03.006]
- Zhang Y, Li QN, Zhou K, Xu Q, Zhang CY. Identification of specific N6-methyladenosine RNA demethylase FTO inhibitors by single-quantum-dot-based FRET nanosensors. Anal Chem 2020;92(20):13936-44. [PMID: 32972135 DOI: 10.1021/acs.analchem.0c02828]
- Huff S, Kummetha IR, Zhang L, Wang L, Bray W, et al. Rational design and optimization of m6A-RNA demethylase FTO inhibitors as anticancer agents. J Med Chem 2022;65(16):10920-37. [PMID: 35939803 DOI: 10.1021/acs.jmedchem.1c02075]
- Selberg S, Yu LY, Bondarenko O, Kankuri E, Seli N, et al. Small-molecule inhibitors of the RNA M6A demethylases FTO potently support the survival of dopamine neurons. Int J Mol Sci 2021;22(9):4537. [PMID: 33926120 DOI: 10.3390/ijms22094537]
- Yang T, Li Z, Chen Y, Feng D, Wang G, et al. DrugSpaceX: a large screenable and synthetically tractable database extending drug space. Nucleic Acids Res 2021;49(D1):D1170-8. [PMID: 33104791 DOI: 10.1093/nar/gkaa920]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J Med Chem 2004;47(7):1739-49. [PMID: 15027865 DOI: 10.1021/jm0306430]
- Ylilauri M, Pentikäinen OT. MMGBSA as a tool to understand the binding affinities of filamin–peptide interactions. J Chem Inf Model 2013;53(10):2626-33. [PMID: 23988151 DOI: 10.1021/ci4002475]
- Gu S, Smith MS, Yang Y, Irwin JJ, Shoichet BK. Ligand strain energy in large library docking. J Chem Inf Model 2021;61(9):4331-41. [PMID: 34467754 DOI: 10.1021/acs.jcim.1c00368]
- Fusani L, Palmer DS, Somers DO, Wall ID. Exploring ligand stability in protein crystal structures using binding pose metadynamics. J Chem Inf Model 2020;60(3):1528-39. [PMID: 31910338 DOI: 10.1021/acs.jcim.9b00843]
- Repasky MP, Murphy RB, Banks JL, Greenwood JR, Tubert-Brohman I, et al. Docking performance of the glide program as evaluated on the Astex and DUD datasets: a complete set of glide SP results and selected results for a new scoring function integrating WaterMap and glide. J Comput Aided Mol Des 2012;26(6):787-99. [PMID: 22576241 DOI: 10.1007/s10822-012-9575-9]
- Clark AJ, Tiwary P, Borrelli K, Feng S, Miller EB, et al. Prediction of protein–ligand binding poses via a combination of induced fit docking and metadynamics simulations. J Chem Theory Comput 2016;12(6):2990-8. [PMID: 27145262 DOI: 10.1021/acs.jctc.6b00201]
- Allegra M, Tutone M, Tesoriere L, Attanzio A, Culletta G, et al. Evaluation of the IKKβ binding of indicaxanthin by induced-fit docking, binding pose metadynamics, and molecular dynamics. Front Pharmacol 2021;12:701568. [PMID: 34566634 DOI: 10.3389/fphar.2021.701568]
- Fu L, Shi S, Yi J, Wang N, He Y, et al. ADMETlab 3.0: an updated comprehensive online ADMET prediction platform enhanced with broader coverage, improved performance, API functionality and decision support. Nucleic Acids Res 2024;52(W1):W422-31. [PMID: 38572755 DOI: 10.1093/nar/gkae236]
- Sunderland EM, Hu XC, Dassuncao C, Tokranov AK, Wagner CC, et al. A review of the pathways of human exposure to poly- and perfluoroalkyl substances (PFASs) and present understanding of health effects. J Expo Sci Environ Epidemiol 2019;29(2):131-47. [PMID: 30470793 DOI: 10.1038/s41370-018-0094-1]
- Tang X, Liu S, Chen D, Zhao Z, Zhou J. The role of the fat mass and obesity-associated protein in the proliferation of pancreatic cancer cells. Oncol Lett 2019;17(2):2473-8. [PMID: 30719115 DOI: 10.3892/ol.2018.9873]
- Niu Y, Lin Z, Wan A, Chen H, Liang H, et al. RNA N6-methyladenosine demethylase FTO promotes breast tumor progression through inhibiting BNIP3. Mol Cancer 2019;18(1):46. [PMID: 30922314 DOI: 10.1186/s12943-019-1004-4]
- Zhang W, Han S, Yuan Y, Xu M, Ding A, et al. FTO knockdown-mediated maturation of miR-383-5p inhibits malignant advancement of pancreatic cancer by targeting ITGA3. Biochem Genet 2024;62(4):2652-66. [PMID: 38001392 DOI: 10.1007/s10528-023-10560-0]
- Su R, Dong L, Li Y, Gao M, Han L, et al. Targeting FTO suppresses cancer stem cell maintenance and immune evasion. Cancer Cell 2020;38(1):79-96.e11. [PMID: 32531268 DOI: 10.1016/j.ccell.2020.04.017]
- Pinto AC, Ades F, de Azambuja E, Piccart-Gebhart M. Trastuzumab for patients with HER2 positive breast cancer: delivery, duration and combination therapies. Breast 2013;22(Suppl 2):S152-5. [PMID: 24074778 DOI: 10.1016/j.breast.2013.07.029]
- Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Cowey CL, et al. Combined nivolumab and ipilimumab or monotherapy in untreated melanoma. N Engl J Med 2015;373(1):23-34. [PMID: 26027431 DOI: 10.1056/NEJMoa1504030]