Multiscale End-point Screening with Extended Tight-binding Hamiltonians
1Faculty of Synthetic Biology, Shenzhen University of Advanced Technology, Shenzhen 518107, China
2College of Physical Science and Technology, Yangzhou University, Yangzhou 225009, China
3Faculty of Biosciences, Taizhou Technician College, Zhejiang 318000, China
aCo-first author.
*Correspondence to: Zhaoxi Sun, Faculty of Synthetic Biology, Shenzhen University of Advanced Technology, Shenzhen 518107, China. E-mail: z.sun@suat-sz.edu.cn
Received: April 20 2025; Revised: June 8 2025; Accepted: July 7 2025; Published Online: July 23 2025
Cite this paper:
Wang X, Li S, Zhang Z et al. Multiscale End-point Screening with Extended Tight-binding Hamiltonians. BIO Integration 2025; 6: 1–11.
DOI: 10.15212/bioi-2025-0071. Available at: https://bio-integration.org/
Download citation
© 2025 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Background: Extended tight-binding (xTB) methods offer a computationally efficient alternative to classical force fields and ab initio quantum methods in modeling molecular systems. In the context of end-point free energy calculations, integrating xTB with implicit solvation models provides a promising route for enhanced accuracy. However, systematic benchmarking of xTB-based protocols remains limited, particularly in diverse host-guest systems.
Methods: We investigated the integration of xTB Hamiltonians (GFN0, GFN1, and GFN2) with post-simulation implicit-solvent models [Poisson−Boltzmann (PB), generalized Born (GB), and the most recent CPCM-X] for end-point free energy calculations. A total of over 250 host-guest complexes were used, covering cucurbiturils, octa acids, and pillararenes. Both single-trajectory and three-trajectory sampling protocols were applied. Entropic contributions were estimated via MM-based normal mode analysis and xTB-based statistical approximations. We evaluated predictive performance using Kendall τ, Pearson r, and predictive index.
Results: The three-trajectory protocol consistently outperformed the single-trajectory counterpart across Hamiltonians and solvent models. Among all configurations, the GFN2-xTB/PB combination showed the best predictive accuracy, although it fell short of the top-performing MM/GBOBCSA ΔG method. Notably, in challenging systems like sulfur-substituted pillararenes, xTB methods exhibited superior performance, whereas MM/GBSA failed due to inadequate error cancellation. The use of CPCM-X did not further enhance accuracy, possibly due to unsuccessful error cancellation.
Conclusions: While MM/GBSA remains the most robust protocol for general use, the GFN2-xTB/PB ΔH method emerges as a viable alternative for cases where MM-based methods perform poorly. These findings highlight the value of xTB-based multiscale approaches for receptor-ligand binding, especially in complex or chemically diverse systems.
Keywords
End-point free energy calculation, extended tight binding, host-guest binding, implicit solvent, multi-trajectory sampling.
Introduction
End-point free energy calculations are based on the principle in which the calculation of the binding strength of a receptor-acceptor complex can be approximated through end-point sampling and post-processing energetic evaluations. End-point sampling neglects the gradual spatial rearrangement of the receptor-acceptor systems and includes only the end-point ensemble (i.e., the bound complex in most cases, and rarely the unbound states) under consideration, whereas post-simulation calculation changes the ensemble, such that the configurational averaging is performed without rigorous reweighting treatment (e.g., from MM explicit-solvent simulations to MM/implicit-solvent single-point calculations). Such free energy techniques are approximated treatments by design, and their success relies on the appropriateness of the approximations used, the accuracy of the Hamiltonians used, and most importantly the cancellation of energetic errors of various origins.
The most widely applied end-point protocol in biomolecular and drug investigations is MM/GBSA with a single-trajectory sampling regime [1–7]. This method samples only the bound-state ensemble with fixed-charge force fields in explicit solvents and post-processes the accumulated snapshots with the same force field in conjunction with a parametrized implicit solvent. This regime is frequently believed to achieve a screening power greater than that of traditional molecular docking but less than that of costlier regimes that rigorously treat the thermodynamic variations during the binding events. Modifications of the naïve single-trajectory MM/GBSA protocol are less frequently used but have been shown to exhibit higher screening power than the commonly used protocol in several cases [8–13]. Among modified regimes, the dielectric constants [14, 15] are frequently varied. Other techniques, such as shifting to higher-level (semi-empirical) QM descriptions, have rarely been explored. The three-trajectory sampling protocol, owing to higher energetic fluctuations, has also been neglected in mainstream applications.
Extended tight-binding (xTB) methods, relatively new semi-empirical QM techniques, serve as a balanced protocol to treat non-covalent interactions in biochemical and biophysical clusters. Their accuracy surpasses that of existing semi-empirical protocols such as PM6-D3H4X [16] and DFTB3 [17, 18] in various energetic benchmarks [19–21]. Although many semi-empirical QM levels (such as AM1, PM6, and DFTB3) have been explored in the end-point modification QM/GBSA, a systematic examination of GFN-xTB implicit-solvent Hamiltonians remains lacking in the approximated screening protocol.
Host-guest complexes with structural features similar to those of receptor-acceptor systems involving biomolecules have broad applications in the drug industry [22, 23]. In practice, their ability to stably coordinate drug-like molecules with tunable strengths makes them promising for controlled release, drug carrier, and reservoir applications [24–26]. Structurally, these macromolecules are formed from several repeating units, and contain a hydrophobic central cavity encapsulating external agents and rims with specific physiochemical features. Despite their relatively simple structural features, calculating host-guest binding affinities remains challenging, even with modern computational chemistry [27–32]. Recent reports on advanced end-point modifications have highlighted the screening power of modified end-point regimes, which has approached and even surpassed that of costlier alchemical methods [8–10, 33]. In terms of the specific end-point modification QM/GBSA, whereas all single-trajectory end-point protocols have exhibited a major failure on the SAMPL9 pillararene dataset, the three-trajectory DFTB/GBSA regime has achieved unexpectedly high performance [8]. Given the similarity of DFTB and GFN-xTB, and the higher energetic accuracy of the latter, we hypothesized that the xTB-based implicit-solvent Hamiltonian might serve as a promising protocol for end-point screening. Therefore, in this work, we present a thorough exploration of the combinations of the xTB levels and implicit-solvent models in end-point screening, by using a series of host-guest datasets as the test bed.
To achieve sufficient coverage of the chemical space and generate a representative evaluation set, we considered several host-guest datasets generated in our previous studies. Macromolecular hosts belonging to three host families: the pumpkin-like cucurbit[n]urils (CBn with seven or eight repeating units), the basket-like octa acids (OA), and pillar[n]arenes (carboxylated WPn chemical modifications with n = 5, 6, 7, and sulfur-substituted SP6). The number of guest molecules binding each host target was unprecedently large: the total number of host-guest pairs exceeded 250. The basic statistics of the test bed are presented in Table 1 and Figure 1. Below, we describe the advanced end-point ranking methods explored in this work, investigation of various settings in calculations, protocol of the best-performing GFN-xTB implicit-solvent protocol, and head-to-head comparison between the advanced regimes and traditional MM/GBSA scoring, to evaluate the practical value of the modified regime.
Figure 1 Distributions of the host-guest binding affinities for each dataset, with the 3D structure of the host target shown in the inset.
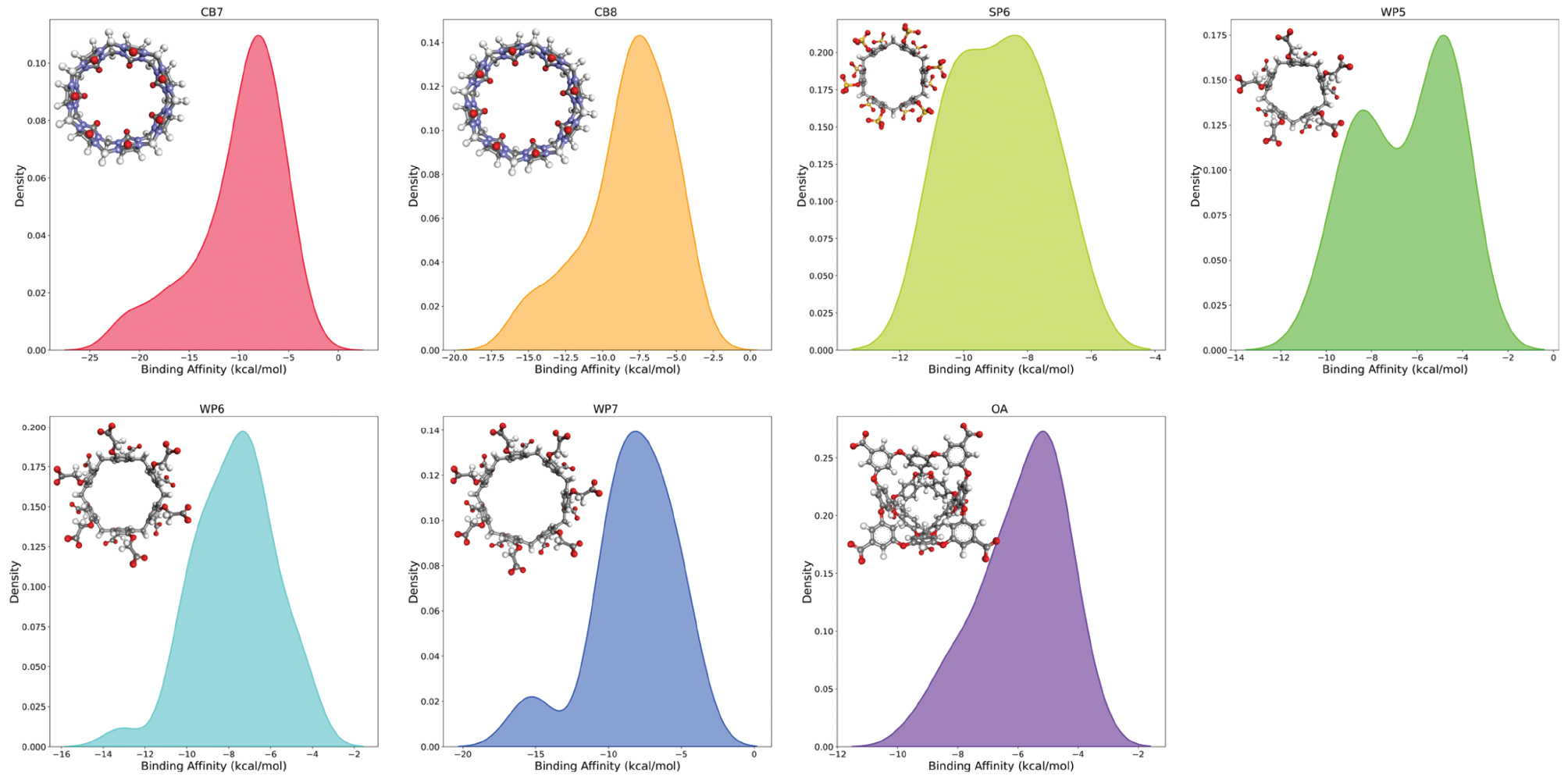
Table 1 Host-guest Datasets Considered Herein. Experimental Affinities are Available in our Previous Articles [34–36]
| Properties | Host | ||||||
|---|---|---|---|---|---|---|---|
| CB7 | CB8 | OA | SP6 | WP5 | WP6 | WP7 | |
| Host Size (Atoms) | 126 | 144 | 184 | 126 | 125 | 150 | 175 |
| Host Repeating Units | 7 | 8 | 4 | 6 | 5 | 6 | 7 |
| Host Net Charge | 0 | 0 | −8 | −12 | −10 | −12 | −14 |
| Guests Binding Host | 88 | 57 | 31 | 11 | 19 | 40 | 23 |
| Affinity Range (kcal/mol) | [−22.15, −2.87] | [−15.81, −3.49] | [−9.37, −3.73] | [−10.87, −6.76] | [−10.16, −3.78] | [−13.14, −4.27] | [−15.73, −4.39] |
Methods
Construction of the end-point ensemble
We directly applied the parameters and configurational ensembles from our previous studies in the current investigation. Basic details regarding the parametrization of all-atom models included RESP [37] charges at HF [38–40]/6-31G* (with structures optimized at B3LYP [41–43]/6-31G*), GAFF derivatives [44] for host and guest molecules (GAFF2 for cucurbiturils and OA, and GAFF for pillararenes, all of which were selected according to recently benchmarked host dynamics) [31, 32, 45], spherical monovalent counter ions (Na+ or Cl−) [46, 47] for neutralization, and TIP3P water [48] with a 15 Å solute-edge distance for solvation. The construction of the bound structure was performed in AutoDock-Vina with the default Vina scoring function [49, 50].
Regarding sampling of the configurational space, starting from the solvated systems (host, guest, and host-guest complex), we relaxed the system with energy minimization, sub-ns constant-volume heating, and 1 ns NPT equilibration, and then sampled each ensemble for at least 200 ns with a 10 ps sampling interval between successive configurations. The unbound host ensemble was sampled for slightly longer than the unbound guest and the bound host-guest systems, this sampling strategy was designed to minimize the overall statistical uncertainty of the three-trajectory end-point free energy estimates based on our previous studies, which showed that the unbound host ensemble exhibited higher energetic fluctuations and was more difficult to sample accurately [8, 9, 34, 45]. The exact sampling lengths for each system have been described in our previous studies [34–36]. The parameters of the molecular simulations included SHAKE constraints [51, 52] on bonds involving hydrogen, a 2 fs time step, Langevin dynamics for temperature regulation at 300 K, isotropic scaling, a Monte Carlo barostat for pressure regulation at 1 atm, an 8 Å real-space cutoff for non-bonded interactions, and PME for long-range electrostatics. The hybrid-precision GPU engine in AMBER [53] was used.
Post-simulation end-point ranking
The end-point estimates of the receptor-acceptor binding strength were formulated as follows:
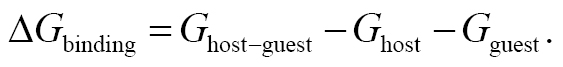 (1)
(1)
Each free energy term, under a force-field framework with decomposable components, was further decomposed to:
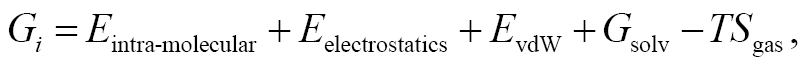 (2)
(2)
where all terms are self-explanatory. The single-trajectory sampling protocol computed all three terms in Eq. (1) with configurations extracted from the bound-state ensemble (i.e., host-guest complex), thus leading to the exact cancellation of the intra-molecular energetics, i.e., the first term of Eq. (2). Consequently, the single-trajectory end-point estimate was computed as the sum of the inter-molecular electrostatics and vdW interactions, the solvation contribution from implicit-solvent models, and the entropic contributions estimated with various approximations (e.g., fluctuations of inter-molecular interactions [54, 55], normal mode [56], or quasi-harmonic analysis [57]). The three-trajectory sampling protocol was used to compute the three energetics with cumulative configurations from individual ensembles (i.e., the configurations from the complex ensemble for Ghost–guest, the host structures from the solvated host ensemble for Ghost, and the guest structures from the guest-only ensemble for Gguest). This treatment had a drawback of enhanced energetic fluctuations, thus hindering convergence to a certain uncertainty level. Consequently, the three-trajectory sampling protocol required longer sampling than the single-trajectory sampling protocol. Our recent studies have indicated the promise of the three-trajectory sampling protocol, which, despite its high computational costs, significantly outperformed the naïve single-trajectory regime in a series of host-guest datasets [8, 9, 34, 35].
In the xTB implicit solvent calculations, the GFN0, GFN1, and GFN2 [19] semi-empirical treatments were combined with analytical polarizable Poisson−Boltzmann (PB) and generalized Born (GB) [58] implicit solvent models. In addition, CPCM-X, a recently developed, more accurate conductor-like polarizable continuum model for semi-empirical methods, was also included in calculations. Because CPCM-X [59] is parametrized for the GFN2-xTB Hamiltonian, for this model, we considered only the GFN2-xTB/CPCM-X combination. Consequently, the scoring methods based on the xTB implicit-solvent treatment included seven protocols. We also considered traditional MM/GBSA estimates for comparison with the xTB implicit-solvent results. For MM/GBSA calculations, we considered the RESP-GAFF(2) description for solutes (host and guest molecules) and GBOBC [60, 61] and GBneck2 [62] for implicit solvents (water). Regarding the entropic contribution, the entropy change was computed with MM/GBHCT [63, 64] by using the standard normal-mode analysis in the MM/GBSA regime, whereas in the xTB implicit-solvent scenario, the entropic contribution was estimated theoretically, with the enthalpic term computed with the modified RRHO regime. Notably, for MM/GBSA calculations, all snapshots from the simulations were included, whereas for xTB implicit-solvent calculations, a further subsampling factor of 30 was used, owing to the increased computational burden associated with elevating the level of theory. The post-simulation xTB implicit-solvent calculations were performed with in-house scripts, whereas the MMPBSA.py [65] script in AMBER was used for ordinary MM/GBSA calculations.
Statistical metrics for quality evaluation
Because the central goal of virtual screening is identifying and ranking top binders to a given target, we considered three correlation metrics: the Kendall τ [66], predictive index (PI) [67], and Pearson r. The Kendall ranking coefficient is used to evaluate the consistency of the predicted rank of binding affinities with respect to the reference experimental rank, a widely applied correlation metric in statistical analysis, whereas PI can be considered an altered version of τ considering the difference between exact values of binding affinities in the experimental reference. Both metrics are ranking coefficients that quantify the ranking power of a given method. In contrast, the Pearson correlation coefficient provides a quantitative estimate of the linear correlation between experimental and computed values and is considered a metric evaluating the scoring power. Consequently, these three metrics were used to evaluate both the ranking and scoring power of a given method. All three metrics ranged from −1 and 1, and a high-performance predictor was expected to have a positive value (close to 1).
Although the above statistical estimators can be used to assess the predictive performance of a method on a specific target, they do not capture a broader picture across multiple datasets. Although weighted sums of these metrics on different datasets could be considered, the final outcome might be substantially influenced by the weighting regime. Therefore, we introduced top N analysis as a more robust and statistically reliable approach to evaluate the consistency and robustness of a given method across multiple targets. We selected the N = 1 scenario (i.e., top 1 analysis) as an illustrative case and explained the numerical details. The top 1 analysis quantified the frequency at which method m was the top-performing method, defined as
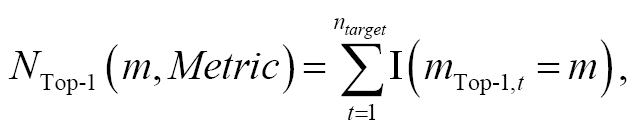 (3)
(3)
where the loop was performed over all host-guest datasets containing ntarget hosts to determine the cumulative host-specific performance. The best method for the host/target t is defined as
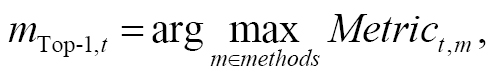 (4)
(4)
with the quality metric being the Kendall, PI, or Pearson correlation coefficient. The NTop 1 count/frequency was bounded by 0 and ntarget. The generalization of the other top N scenarios was straightforward.
Results and discussion
Entropic contribution at MM and xTB levels
In end-point free energy calculations with either the traditional MM/GBSA or more advanced modifications (e.g., dielectric-constant variable), the entropic contribution is often computed with the MM/GBSA-based Hamiltonian, i.e., MM/GBHCT or gas-phase MM. This approximated treatment is adopted for several reasons. First, the use of this approximated treatment is largely driven by the high computational cost of the normal-mode procedure. If replacement of the Hamiltonian handling the solute molecules with another is expected, e.g., from MM to DFTB (leading to QM/GBSA treatment), the computational cost of the normal-mode analysis for the same system would markedly increase. In this case, combining the enthalpic component computed with the changed Hamiltonian with the entropic contribution computed at a given level appears to be feasible and acceptable. In addition, given the high cost of the normal-mode analysis, in most cases, only a small portion of the accumulated simulation snapshots (e.g., ~20 configurations or even none) are included in calculations for protein-ligand and protein-protein binding. Second, the most important and direct reason for the frequent application of MM/GBHCT or gas-phase MM in normal-mode analysis is the limited flexibility of the mainstream implementation MMPBSA.py [65]. This AMBER-based program/script merely supports the two aforementioned levels of theory. In modified end-point workflows, limiting the level of theory for entropic contribution does not appear to be sufficiently accurate. Third, the Hamiltonian dependence of the entropy change is often believed to be small, but the practical influence of such systematic bias remains unexplored. The combination of energetics at different levels of theory would introduce systematic errors, which might be particularly pronounced for multiscale QM/GBSA and its variants. Therefore, in the current xTB implicit-solvent investigation, we first determined the validity of this approximation and quantified the magnitude of the errors introduced.
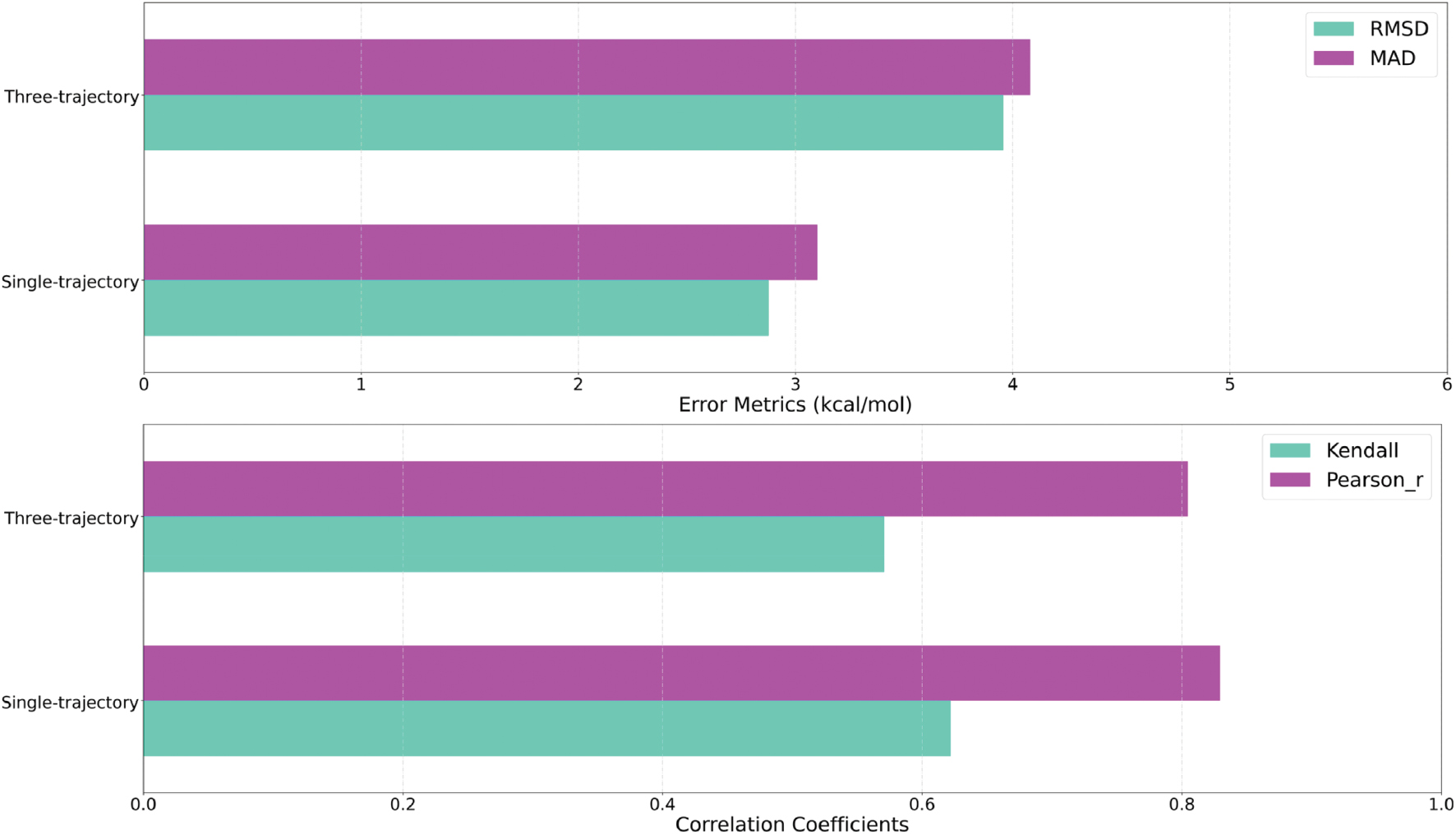
We compared the MM-based and GFN0-xTB/GB entropy estimates in both single- and three-trajectory sampling protocols for all host-guest pairs (Figure 2). Four statistical metrics were considered: root-mean-square deviation and mean absolute deviation for measuring the deviations of absolute values, and the Kendall τ [66] and Pearson r for evaluating the consistency of the ranks of the two entropy changes and the linear correlation. The calculation procedure simply involved placing (MM and xTB-based) entropic contributions into two columns and using a simple Python script for comparison. According to error metrics (root-mean-square deviation ~4.5 kcal/mol and mean absolute deviation ~3.5 kcal/mol), the absolute values of the entropic contribution exhibited noticeable differences across different levels of theory. However, according to the large Kendall τ ranking coefficient of ~0.6, the relative sizes of the entropic contributions in different host-guest pairs were largely consistent. Similarly, the high Pearson correlation coefficient of ~0.8 suggested a good linear correlation between MM- and xTB-based entropic contributions. Overall, whereas the entropy changes after binding, computed under different Hamiltonians, exhibited correlations, non-negligible discrepancies were observed between the absolute values of the MM-based normal-mode and the xTB implicit-solvent estimates. Because the statistical uncertainties of free energy calculations are often expected to be minimal (e.g., thermal energy kBT or chemical accuracy of 1 kcal/mol), and smaller than the Hamiltonian dependence of the entropic term, the above analyses underscored that approximating the entropic contribution in modified end-point procedures with another (e.g., MM/GBHCT or gas-phase MM) would introduce statistically significant systematic biases. This behavior was not limited to the xTB-based results but should be generally applicable to other QM/GBSA methods (e.g., DFTB/GBSA).
Figure 2 Statistical metrics for evaluating the consistency and deviations between the MM- and xTB-based entropy changes for single-trajectory and three-trajectory sampling protocols. The statistics were computed for all host-guest pairs.
Robust xTB implicit-solvent protocols across host families
We next examined the apparent performance of xTB implicit-solvent methods. The aim of end-point screening is to rank the strength of binding of different guests to a given host, and the screening power is often estimated by ranking power (Kendall τ [66] and PI [67]) and scoring power (Pearson r). The ranking and scoring statistics in all datasets are shown in Figure S1–S3. The varied end-point parameters included the inclusion/exclusion of the entropic contribution, the sampling protocol (single- or three-trajectory), the implicit-solvent model (PB, GB, and CPCM-X), and the xTB variants (GFN0-xTB, GFN1-xTB, and GFN2-xTB). Because our goal was to identify a robust approach that performed consistently well across most datasets, we did not focus on a single dataset but instead explored general trends and behaviors of the xTB implicit-solvent treatments.
Notably, given the same Hamiltonian (xTB level, implicit solvent model, and entropic contribution), the three-trajectory sampling protocol appeared to perform consistently better than the frequently used single-trajectory sampling protocol. This behavior under the xTB implicit-solvent scoring was consistent with our previous observations in MM-based regimes (MM/GBSA and MM/PBSA) and other semi-empirical QM/GBSA modifications [8, 9, 68]. Therefore, the superiority of the three-trajectory regime should be fairly general in end-point screening. Another interesting observation was the unexpected underperformance of the GFN2-xTB/CPCM-X scheme. This Hamiltonian, the most recent parametrization of the xTB implicit-solvent Hamiltonians, is considered the highest-level protocol and has been found to produce more accurate energetics in several benchmarks [59]. However, these findings have not led to better screening power for host-guest complexes, possibly because of the unsuccessful cancellation of errors from various origins/approximations.
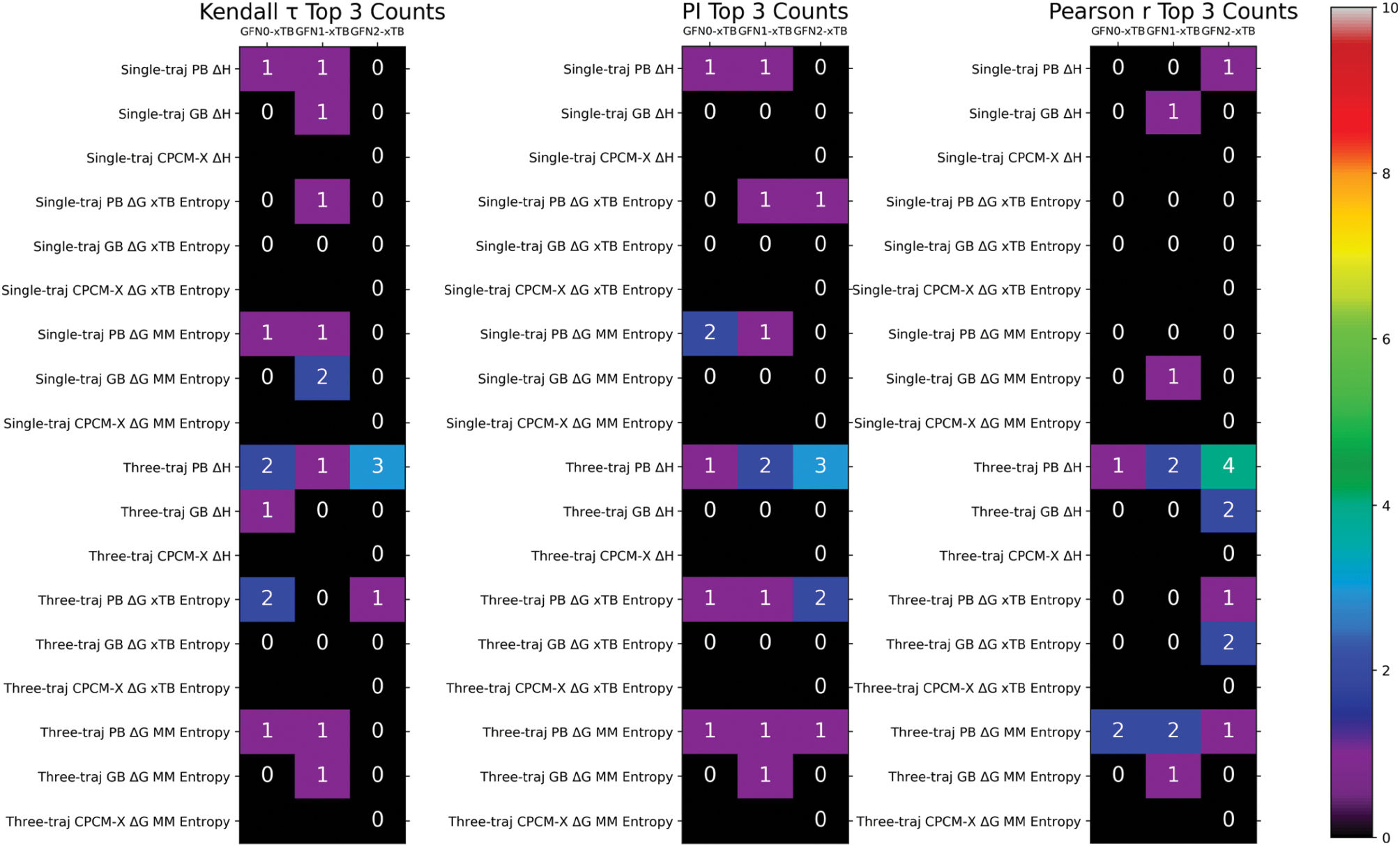
To obtain more general insights from the statistical analyses, we selected the top N methods according to each of the three correlation coefficients for all host-guest datasets and generated count maps to identify top-performing methods that were robust across all host families. The top 3 frequency analyses based on the Kendall τ, PI, and Pearson r are presented in Figure 3. Whereas the best-performing protocols varied with the statistical metrics, the three-trajectory GFN2-xTB/PB ΔH regime consistently showed the best performance in all host-guest datasets. Similar conclusions were reached with the other frequency maps (top 5 in Figure S4 and top 7 in Figure S5).
Figure 3 Top N analysis with N = 3. The N top-performing methods in each host-guest dataset were extracted, and a frequency analysis was performed. The most robust technique had the largest number of occurrences in the heatmap. The top 5 and top 7 analyses provided in the supporting information indicated similar trends.
MM/GBSA vs GFN2-xTB/PB: practical value of xTB implicit-solvent postprocessing
The best-performing and most robust xTB implicit-solvent protocol was demonstrated to be the three-trajectory GFN2-xTB/PB ΔH regime. To illustrate the practical value of using the xTB implicit-solvent regime in end-point screening, we compared its performance statistics with those of the traditional MM/GBSA regime (Figure 4). Across seven host-guest datasets, the end-point method with the highest robustness with top 1 counts of ~3 for all three correlation metrics was the three-trajectory MM/GBOBCSA ΔG protocol. In comparison, the other two protocols, three-trajectory MM/GBneck2SA ΔG and three-trajectory GFN2-xTB/PB ΔH, each attained a top 1 count of 2. These findings suggested that the upgraded xTB implicit-solvent treatment, despite increases in energetic accuracy, unfortunately did not lead to more robust screening power in host-guest binding. The end-point protocol that achieved the best error cancellation and consequently screening accuracy was the three-trajectory MM/GBOBCSA ΔG regime.
Figure 4 Comparison between the performance statistics of the traditional MM/GBSA and the GFN2-xTB/PB ΔH regimes: A) Kendall τ, B) PI, and C) Pearson r. Magenta circles indicate the best-performing protocol for each dataset.
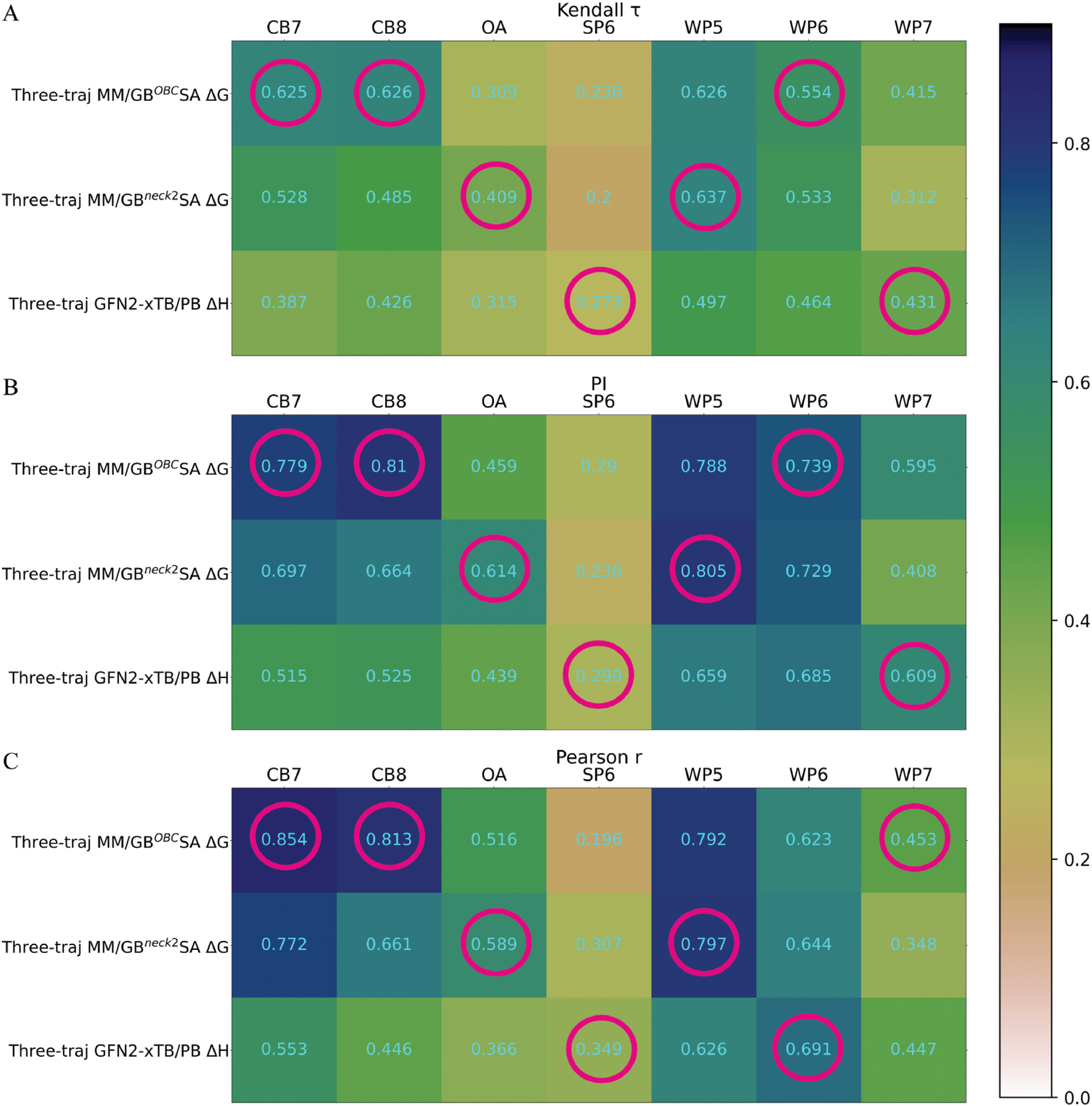
Although the GFN2-xTB/PB ΔH regime was not the best or most robust protocol, it demonstrated an interesting behavior: the targets where the xTB protocol performed best were often the same cases where MM/GBSA methods failed. This aspect was particularly evident for the SP6 host, for which both MM/GBSA-based regimes severely failed. This performance gap may be attributed to less effective error cancellation in the MM/GBSA regime compared to the xTB-based approach for the MM/GBSA regime, and indicated that the elevation of the level of theory might be a potentially useful option in such difficult scenarios. Notably, a similar observation has also been reported in a publicly accessible dataset, SAMPL9 carboxylated pillararene [69]. In that case, the three-trajectory DFTB/GBSA regime achieved unexpectedly high correlation coefficients when compared to experimental binding affinities and performed better than costlier alchemical free energy calculations [8]. Therefore, although we recommend application of the three-trajectory MM/GBOBCSA ΔG regime for host-guest binding in most cases, the three-trajectory GFN2-xTB/PB ΔH option is a recommended alternative that might be applied in difficult scenarios.
Concluding remarks
Herein, we presented an extensive exploration of the xTB implicit-solvent Hamiltonian in end-point free energy calculation of host-guest binding. Using an unprecedently comprehensive test bed containing seven macromolecular hosts from three commonly used host families (cucurbiturils, OA, and pillararenes) and more than 250 host-guest pairs with diverse features, we thoroughly benchmarked all possible parameter combinations in terms of the sampling protocol (the commonly used single-trajectory and the less commonly applied three-trajectory sampling protocols), xTB parametrization (GFN0-xTB, GFN1-xTB, or GFN2-xTB), implicit-solvent model (PB, GB, or the latest CPCM-X), and the method of computing entropic contribution (using the same theoretical level for both enthalpic and entropic contributions, the MM/GBHCT level default in the commonly used MMPBSA.py script, or simply neglected). Several conclusions are presented below:
- Entropic contributions computed with different Hamiltonians exhibited statistically significant differences. Therefore, combining the enthalpic contribution estimated at a selected level with the entropy change at another level can introduce non-negligible systematic biases.
- Among the many xTB implicit-solvent end-point protocols, the parameter combination achieving the highest screening power across all datasets was the three-trajectory GFN2-xTB/PB ΔH method.
- Unfortunately, upgrading the implicit-solvent model to CPCM-X produced more accurate energetics than PB and GB in several benchmarks but did not increase the end-point screening accuracy, possibly because of less successful error cancellation.
- Even the best xTB-based option, the three-trajectory GFN2-xTB/PB ΔH method, did not achieve better robustness than the best MM/GBSA regime, three-trajectory MM/GBOBCSA ΔG. Therefore, in most cases, we recommend the three-trajectory MM/GBOBCSA ΔG as the default option for host-guest screening.
- Interestingly, in some scenarios, the MM/GBSA-based techniques severely failed (e.g., SP6 host-guest complexes). In these difficult cases, shifting to the three-trajectory GFN2-xTB/PB ΔH achieved improvements; therefore, the xTB implicit-solvent regime is an alternative worthy of testing when MM-based techniques fail.
The current work highlighted the potential utility of the xTB implicit-solvent end-point screening in host-guest systems. Because of the commonly acknowledged similarities between host-guest and protein-ligand complexes, these xTB-based protocols might also be applicable to protein-ligand binding, thus providing higher-level protocols in the pool of end-point free energy tools.
Data availability statement
All software used in this work can be accessed freely for academic use, and the parameter files used in molecular simulations are available in our previous studies [34–36].
Ethics statement
No direct interactions with human or animal subjects were involved. Therefore, ethical approval and informed consent were not required.
Author contributions
Xiaohui Wang: Data curation, Formal analysis, Funding acquisition, Investigation, Methods, Resources, Software, Validation, Writing—review & editing.
Sai Li: Formal analysis, Funding acquisition, Investigation, Resources, Software, Writing—review & editing.
Zuo-yuan Zhang: Formal analysis, Funding acquisition, Investigation, Resources, Software, Writing—review & editing.
Linqiong Qiu: Formal analysis, Funding acquisition, Investigation, Resources, Software, Writing—review & editing.
Zhaoxi Sun: Conceptualization, Data curation, Formal analysis, Funding acquisition, Investigation, Methods, Project administration, Resources, Software, Supervision, Validation, Visualization, Writing–original draft, Writing—review & editing.
Acknowledgements
This work was supported by the Beijing Natural Science Foundation (grant No. 7224357) and National Natural Science Foundation of China (grant Nos. 22250710136 and 92270001). We thank the anonymous reviewers for valuable comments and critical reading.
Conflict of interest statement
The authors declare that there are no conflicts of interest.
Supporting information description
The ranking and scoring statistics in all host-guest datasets (Kendall τ in Figure S1, PI in Figure S2, and Pearson r in Figure S3), the top N frequency maps (top 5 maps in Figure S4 and top 10 maps in Figure S5) are provided in the supporting information. Supplementary Material can be downloaded from https://bio-integration.org/wp-content/uploads/2025/07/bioi20250071_Supplemental.pdf.
Graphical abstract

Highlights
- Extended tight-binding (xTB) methods were integrated with end-point free energy calculations to evaluate receptor-ligand binding.
- Both single-trajectory and three-trajectory sampling protocols were benchmarked by using various xTB Hamiltonians and implicit solvent models.
- The study used diverse host-guest datasets featuring cucurbiturils, octa acids, and pillararenes paired with multiple guest molecules.
- The three-trajectory GFN2-xTB/PB approach delivered consistently strong performance across various host families.
- Whereas MM/GBSA generally had better performance, xTB methods provided advantages in challenging cases, such as sulfur-substituted hosts.
In brief
This study comprehensively evaluated xTB implicit-solvent Hamiltonians in end-point free energy calculations for host-guest binding. Using more than 250 host-guest complexes spanning seven hosts comprising cucurbiturils, octa acids, or pillararenes, the authors systematically assessed combinations of sampling protocols, xTB levels (GFN0–2), solvent models (PB, GB, and CPCM-X), and entropy treatments. The three-trajectory GFN2-xTB/PB ΔH method emerged as the most reliable xTB-based protocol but generally fell short of the best-performing MM/GBSA approach (three-trajectory MM/GBOBCSA ΔG). Notably, xTB methods outperformed MM/GBSA in specific challenging cases, such as sulfur-containing systems, thus highlighting their value as alternative tools when MM-based models struggle. These findings underscore the promise of xTB end-point protocols in host-guest and potentially protein-ligand binding applications.
References
- Sun J, Liu X, Zhang S, Li M, Zhang Q, et al. Molecular insights and optimization strategies for the competitive binding of engineered ACE2 proteins: a multiple replica molecular dynamics study. Phys Chem Chem Phys 2023;25:28479-96. [DOI: 10.1039/D3CP03392A]
- Meng L, Xinguo L, Shaolong Z, Jiahao S, Qinggang Z, et al. Selective mechanism of inhibitors to two bromodomains of BRD4 revealed by multiple replica molecular dynamics simulations and free energy analyses. Chin J Chem Phys 2023;36(6):725-39. [DOI: 10.1063/1674-0068/cjcp2208126]
- Yang J, Chen L, Huang X, Zhao B, Wang R. Binding interactions of EDCs to human estrogen-related receptor gamma deciphered by multiple molecular dynamics and energy calculations. Int J Quantum Chem 2024;124:e27333. [DOI: 10.1002/qua.27333]
- Zhou Y, Liu X, Zhang Y, Peng L, Zhang JZ. Residue-specific free energy analysis in ligand bindings to JAK2. Mol Phys 2018;116:2633-41. [DOI: 10.1080/00268976.2018.1442596]
- Patil VM, Gupta SP, Masand N, Balasubramanian K. Experimental and computational models to understand protein-ligand, metal-ligand and metal-DNA interactions pertinent to targeted cancer and other therapies. Eur J Med Chem Rep 2024;10(51):100133. [DOI: 10.1016/j.ejmcr.2024.100133]
- Pan Y, Zhao C, Fu W, Yang S, Lv S. Comparative analysis of structural dynamics and allosteric mechanisms of RecA/Rad51 family proteins: Integrated atomistic MD simulation and network-based analysis. Int J Biol Macromol 2024;261(2):129843. [DOI: 10.1016/j.ijbiomac.2024.129843]
- Yu Z, Wang Z, Cui X, Cao Z, Zhang W, et al. Conformational states of the GDP- and GTP-bound HRAS affected by A59E and K117R: an exploration from Gaussian accelerated molecular dynamics. Molecules 2024;29:645. [PMID: 38338389 DOI: 10.3390/molecules29030645]
- Wang X, Wang M, Sun Z. Comprehensive evaluation of end-point free energy techniques in carboxylated-pillar[6]arene host-guest binding: IV. The QM treatment, GB models and the multi-trajectory extension. Liquids 2023;3:426-39. [DOI: 10.3390/liquids3040027]
- Liu X, Zheng L, Qin C, Yalong C, Zhang JZ, et al. Comprehensive evaluation of end-point free energy techniques in carboxylated-pillar[6]arene host-guest binding: III. Force-field comparison, three-trajectory realization and further dielectric augmentation. Molecules 2023;28:2767. [DOI: 10.3390/molecules28062767]
- Liu X, Zheng L, Cong Y, Gong Z, Yin Z, et al. Comprehensive evaluation of end-point free energy techniques in carboxylated-pillar[6]arene host-guest binding: II. Regression and dielectric constant. J Comput Aided Mol Des 2022;36:879-94. [PMID: 36394776 DOI: 10.1007/s10822-022-00487-w]
- Liu X, Peng L, Zhang JZ. Accurate and efficient calculation of protein–protein binding free energy-interaction entropy with residue type-specific dielectric constants. J Chem Inf Model 2018;59:272-81. [PMID: 30431271 DOI: 10.1021/acs.jcim.8b00248]
- Genheden S, Ryde U. The MM/PBSA and MM/GBSA methods to estimate ligand-binding affinities. Expert Opin Drug Discov 2015;10:449-61. [PMID: 25835573 DOI: 10.1517/17460441.2015.1032936]
- Genheden S, Kuhn O, Mikulskis P, Hoffmann D, Ryde U. The normal-mode entropy in the MM/GBSA method: effect of system truncation, buffer region, and dielectric constant. J Chem Inf Model 2012;52:2079-88. [PMID: 22817270 DOI: 10.1021/ci3001919]
- Wang E, Weng G, Sun H, Du H, Zhu F, et al. Assessing the performance of the MM/PBSA and MM/GBSA methods. 10. Impacts of enhanced sampling and variable dielectric model on protein–protein interactions. Phys Chem Chem Phys 2019;21:18958-69. [DOI: 10.1039/C9CP04096J]
- Yang T, Wu JC, Yan C, Wang Y, Luo R, et al. Virtual screening using molecular simulations. Proteins 2011;79:1940-51. [PMID: 21491494 DOI: 10.1002/prot.23018]
- Brahmkshatriya PS, Dobeš P, Fanfrlik J, Rezáç J, Paruch K, et al. Quantum mechanical scoring: structural and energetic insights into cyclin-dependent kinase 2 inhibition by pyrazolo [1, 5-a] pyrimidines. Curr Comput Aided Drug Des 2013;9:118-29. [PMID: 23157414 DOI: 10.2174/1573409911309010011]
- Gaus M, Goez A, Elstner M. Parametrization and benchmark of DFTB3 for organic molecules. J Chem Theory Comput 2013;9:338-54. [PMID: 26589037 DOI: 10.1021/ct300849w]
- Gaus M, Cui Q, Elstner M. DFTB3: Extension of the self-consistent-charge density-functional tight-binding method (SCC-DFTB). J Chem Theory Comput 2011;7:931-48. [PMID: 23204947 DOI: 10.1021/ct100684s]
- Bannwarth C, Ehlert S, Grimme S. GFN2-xTB–An accurate and broadly parametrized self-consistent tight-binding quantum chemical method with multipole electrostatics and density-dependent dispersion contributions. J Chem Theory Comput 2019;15:1652-71. [PMID: 30741547 DOI: 10.1021/acs.jctc.8b01176]
- Grimme S, Bannwarth C, Shushkov P. A robust and accurate tight-binding quantum chemical method for structures, vibrational frequencies, and noncovalent interactions of large molecular systems parametrized for all spd-block elements (Z = 1–86). J Chem Theory Comput 2017;13:1989-2009. [PMID: 28418654 DOI: 10.1021/acs.jctc.7b00118]
- Spicher S, Grimme S. Efficient computation of free energy contributions for association reactions of large molecules. J Phys Chem Lett 2020;11:6606-11. [PMID: 32787231 DOI: 10.1021/acs.jpclett.0c01930]
- Masson E, Ling X, Joseph R, Kyeremeh-Mensah L, Lu X. Cucurbituril chemistry: a tale of supramolecular success. RSC Adv 2012;2:1213-47. [DOI: 10.1039/C1RA00768H]
- Kim K, Selvapalam N, Oh DH. Cucurbiturils–a new family of host molecules. J Incl Phenom Macrocycl Chem 2004;50:31-6. [DOI: 10.1007/s10847-004-8835-7]
- Corma A, García H, Montes-Navajas P, Primo A, Calvino JJ, et al. Gold nanoparticles in organic capsules: a supramolecular assembly of gold nanoparticles and cucurbituril. Chem Eur J 2007;13:6359-64. [PMID: 17497621 DOI: 10.1002/chem.200601900]
- Saluja V, Sekhon BS. Calixarenes and cucurbiturils: pharmaceutial and biomedical applications. J Pharm Edu Res 2013;4:16.
- Yahiaoui K, Seridi L, Mansouri K. Temozolomide binding to Cucurbit[7]uril: QTAIM, NCI-RDG and NBO analyses. J Incl Phenom Macrocycl Chem 2021;99:61-77. [DOI: 10.1007/s10847-020-01027-5]
- Procacci P, Guarnieri G. SAMPL9 blind predictions using nonequilibrium alchemical approaches. J Chem Phys 2022;156:164104. [PMID: 35490003 DOI: 10.1063/5.0086640]
- Procacci P, Guarrasi M, Guarnieri G. SAMPL6 host–guest blind predictions using a non equilibrium alchemical approach. J Comput Aided Mol Des 2018;32:965-82. [PMID: 30128927 DOI: 10.1007/s10822-018-0151-9]
- Eken Y, Patel P, Díaz T, Jones MR, Wilson AK. SAMPL6 host–guest challenge: binding free energies via a multistep approach. J Comput Aided Mol Des 2018;32:1097-115. [PMID: 30225724 DOI: 10.1007/s10822-018-0159-1]
- Sun Z, Zheng L, Wang K, Huai Z, Liu Z. Primary vs secondary: directionalized guest coordination in β-cyclodextrin derivatives. Carbohydr Polym 2022;297:120050. [DOI: 10.1016/j.carbpol.2022.120050]
- Sun Z, Huai Z, He Q, Liu Z. A general picture of cucurbit[8]uril host–guest binding. J Chem Inf Model 2021;61:6107-34. [PMID: 34818004 DOI: 10.1021/acs.jcim.1c01208]
- Sun Z, He Q, Gong Z, Kalhor P, Huai Z, et al. A general picture of cucurbit[8]uril host-guest binding: recalibrating bonded interactions. Molecules 2023;28:3124. [PMID: 37049887 DOI: 10.3390/molecules28073124]
- Liu X, Zheng L, Qin C, Zhang JZH, Sun Z. Comprehensive evaluation of end-point free energy techniques in carboxylated-pillar[6]arene host-guest binding: I. Standard procedure. J Comput Aided Mol Des 2022;36:735-52. [PMID: 36136209 DOI: 10.1007/s10822-022-00475-0]
- Wang X, Yang H, Wang M, Huai Z, Sun Z. Virtual screening of cucurbituril host-guest complexes: large-scale benchmark of end-point protocols under MM and QM hamiltonians. J Mol Liq 2024;407:125245. [DOI: 10.1016/j.molliq.2024.125245]
- Wang X, Huai Z, Zheng L, Liu M, Sun Z. A benchmark test of high-throughput atomistic modeling for octa acid host-guest complexes. Liquids 2024;4(3):485-504. [DOI: 10.3390/liquids4030027]
- Sun Z. A testing bed for computational modelling of host-guest binding. ChemRxiv 2025. [DOI: 10.26434/chemrxiv-2025-5w068]
- Bayly CI, Cieplak P, Cornell W, Kollman PA. A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: the RESP model. J Phys Chem 1992;97:10269-80. [DOI: 10.1021/j100142a004]
- Mcweeny R, Diercksen G. Self-consistent perturbation theory. II. Extension to open shells. J Chem Phys 1968;49:4852-6. [DOI: 10.1063/1.1669970]
- Pople JA, Nesbet RK. Self-consistent orbitals for radicals. J Chem Phys 1954;22:571-2. [DOI: 10.1063/1.1740120]
- Roothaan CCJ. New developments in molecular orbital theory. Rev Mod Phys 1951;23:69-89. [DOI: 10.1103/RevModPhys.23.69]
- Hertwig RH, Koch W. On the parameterization of the local correlation functional. What is Becke-3-LYP? Chem Phys Lett 1997;268:345-51. [DOI: 10.1016/S0009-2614(97)00207-8]
- Becke AD. Density-functional thermochemistry. IV. A new dynamical correlation functional and implications for exact-exchange mixing. J Chem Phys 1996;104:1040-6. [DOI: 10.1063/1.470829]
- Stephens PJ, Devlin FJ, Chabalowski CF, Frisch MJ. Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields. J Phys Chem 1994;98:11623-7. [DOI: 10.1021/j100096a001]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem 2004;25:1157-73. [PMID: 15116359 DOI: 10.1002/jcc.20035]
- Wang X, Huai Z, Sun Z. Host dynamics under general-purpose force fields. Molecules 2023;28:16. [PMID: 37630194 DOI: 10.3390/molecules28165940]
- Joung IS, Cheatham TE III. Determination of alkali and halide monovalent ion parameters for use in explicitly solvated biomolecular simulations. J Phys Chem B 2008;112:9020-41. [PMID: 18593145 DOI: 10.1021/jp8001614]
- Joung IS, Cheatham TE. Molecular dynamics simulations of the dynamic and energetic properties of alkali and halide ions using water-model-specific ion parameters. J Phys Chem B 2009;113:13279-90. [DOI: 10.1021/jp902584c]
- Price DJ, Brooks CL III. A modified TIP3P water potential for simulation with Ewald summation. J Chem Phys 2004;121:10096-103. [DOI: 10.1063/1.1808117]
- Eberhardt J, Santos-Martins D, Tillack AF, Forli S. AutoDock Vina 1.2.0: new docking methods, expanded force field, and python bindings. J Chem Inf Model 2021;61:3891-8. [PMID: 34278794 DOI: 10.1021/acs.jcim.1c00203]
- Nguyen NT, Nguyen TH, Pham TNH, Huy NT, Bay MV, et al. Autodock Vina adopts more accurate binding poses but Autodock4 forms better binding affinity. J Chem Inf Model 2020;60:204-11. [PMID: 31887035 DOI: 10.1021/acs.jcim.9b00778]
- Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical integration of the cartesian equations of motion of a system with constraints: molecular dynamics of n-alkanes. J Comput Phys 1977;23:327-41. [DOI: 10.1016/0021-9991(77)90098-5]
- Miyamoto S, Kollman PA. Settle: an analytical version of the SHAKE and RATTLE algorithm for rigid water models. J Comput Chem 1992;13:952-62. [DOI: 10.1002/jcc.540130805]
- Case DA, Cheatham TE, Tom D, Holger G, Luo R, et al. The Amber biomolecular simulation programs. J Comput Chem 2005;26:1668-88. [PMID: 16200636 DOI: 10.1002/jcc.20290]
- Sun Z, Yan YN, Yang M, Zhang JZ. Interaction entropy for protein-protein binding. J Chem Phys 2017;146:124124. [PMID: 28388125 DOI: 10.1063/1.4978893]
- Yan Y, Yang M, Ji CG, Zhang JZH. Interaction entropy for computational alanine scanning. J Chem Inf Model 2017;57:1112-22. [PMID: 28406301 DOI: 10.1021/acs.jcim.6b00734]
- Case DA. Normal mode analysis of protein dynamics. Curr Opin Struct Biol 2010;4:285-90. [DOI: 10.1016/S0959-440X(94)90321-2]
- Karplus M, Kushick JN. Method for estimating the configurational entropy of macromolecules. Macromolecules 1981;14:325-32. [DOI: 10.1021/ma50003a019]
- Ehlert S, Stahn M, Spicher S, Grimme S. Robust and efficient implicit solvation model for fast semiempirical methods. J Chem Theory Comput 2021;17:4250-61. [DOI: 10.1021/acs.jctc.1c00471]
- Stahn M, Ehlert S, Grimme S. Extended conductor-like polarizable continuum solvation model (CPCM-X) for semiempirical methods. J Phys Chem A 2023;127:7036-43. [DOI: 10.1021/acs.jpca.3c04382]
- Onufriev A, Bashford D, Case DA. Exploring protein native states and large-scale conformational changes with a modified generalized born model. Proteins 2004;55:383-94. [PMID: 15048829 DOI: 10.1002/prot.20033]
- Feig M, Onufriev A, Lee MS, Im W, Case DA, et al. Performance comparison of generalized born and Poisson methods in the calculation of electrostatic solvation energies for protein structures. J Comput Chem 2004;25:265-84. [PMID: 14648625 DOI: 10.1002/jcc.10378]
- Hai N, Pérez A, Bermeo S, Simmerling C. Refinement of generalized born implicit solvation parameters for nucleic acids and their complexes with proteins. J Chem Theory Comput 2015;11:3714. [DOI: 10.1021/acs.jctc.5b00271]
- Hawkins GD, Cramer CJ, Truhlar DG. Parametrized models of aqueous free energies of solvation based on pairwise descreening of solute atomic charges from a dielectric medium. J Phys Chem 1996;100:19824-39. [DOI: 10.1021/jp961710n]
- Hawkins GD, Cramer CJ, Truhlar DG. Pairwise solute descreening of solute charges from a dielectric medium. Chem Phys Lett 1995;246:122-9. [DOI: 10.1016/0009-2614(95)01082-K]
- Miller BR, McGee TD, Swails JM, Homeyer N, Gohlke H, et al. MMPBSA.py: an efficient program for end-state free energy calculations. J Chem Theory Comput 2012;8:3314-21. [DOI: 10.1021/ct300418h]
- Kendall MG. A new measure of rank correlation. Biometrika 1938;30:81-93. [DOI: 10.2307/2332226]
- Pearlman DA, Charifson PS. Are free energy calculations useful in practice? A comparison with rapid scoring functions for the p38 MAP kinase protein system. J Med Chem 2001:44:3417-23. [DOI: 10.1021/jm0100279]
- Liu X, Zheng L, Qin C, Li Y, Zhang JZH, et al. Screening power of end-point free-energy calculations in cucurbituril host–guest systems. J Chem Inf Model 2023;63:6938-46. [PMID: 37908066 DOI: 10.1021/acs.jcim.3c01356]
- The SAMPL9 blind prediction challenges for computational chemistry. Available at: https://github.com/samplchallenges/SAMPL9.