Ferroptosis Resistance in Cancer: An Emerging Crisis of New Hope
1School of Pharmaceutical Sciences (Shenzhen), Sun Yat-sen University, Shenzhen, 518100, China
2Department of Pathology, The Eighth Affiliated Hospital, Sun Yat-sen University, Shenzhen, 518033, China
*Correspondence to: Zhe Wang and Junqing Wang E-mail: Wangzh379@mail.sysu.edu.cn (Zhe Wang) and wangjunqing@mail.sysu.edu.cn (Junqing Wang)
Received: October 30 2020; Revised: November 16 2020; Accepted: December 18 2020; Published Online: January 7 2021
Cite this paper:
Daiyun Xu, Yonghui Lü, Yongxiao Li, Shengbin Li, Zhe Wang and Junqing Wang. Ferroptosis Resistance in Cancer: An Emerging Crisis of New Hope. BIO Integration 2021; 2(1): 22–28.
DOI: 10.15212/bioi-2020-0039. Available at: https://bio-integration.org/
Download citation 
© 2021 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Ferroptosis, a new mode of nonapoptotic cell death, is increasingly recognized as a new hope in overcoming resistance to chemotherapy in cancer. Both canonical and noncanonical pathways can trigger ferroptosis execution via an iron-dependent lethal lipid peroxidation manner. However, growing evidence has shown that some cancer cells can survive ferroptotic stress through metabolic remodeling as regards iron metabolism, anti-oxidative systems, and lipid metabolism. In addition to the well-known roles of the XC−/glutathione/glutathione peroxidase 4 (XC–/GSH/GPX4) axis in blocking ferroptosis, several recently identified pathways, including the Mevalonate-ferroptosis suppressor protein 1 (MVA-FSP1) axis, the GTP cyclohydrolase 1-Tetrahydrobiopterin (GCH1-BH4) axis, the peroxisome-ether-phospholipid axis, the acyl-CoA synthetase long-chain family member 3-monounsaturated fatty acids (ACSL3-MUFA) axis, and the Liver kinase B1-AMP-activated protein kinase (LKB1-AMPK) axis, can negatively regulate susceptibility to ferroptosis. Prominin-2, a newly identified ferroptosis-modulating protein, also drives cancer cells to escape from ferroptosis induction. These findings collectively led to major challenges and opportunities in the development of novel therapies that target the ferroptosis resistance of cancer cells.
Significance
Ferroptosis is a lethal consequence of accumulated lipid peroxidation catalyzed by ferrous iron and oxygen. This unique cell death process appears to involve many diseases, such as neurodegeneration, ischemia/reperfusion injury, kidney disease, and a druggable target in therapy-resistant cancers. There is great expectation of being able to exploit ferroptosis for the treatment of as yet incurable diseases. However, the state of ferroptosis susceptibility is linked to various regulation pathways. This perspective aims to integrate the current understanding of signaling mechanisms for ferroptotic defenses and facilitates movement toward novel cancer therapeutic strategies.
Introduction
The unregulated proliferation, metastasis, and chemoresistance in cancer progression are pressing challenges in cancer treatment. This severe outcome primarily arises from intrinsic insensitivity or adaptive evasion of apoptosis [1, 2]. Ferroptosis is a recently discovered new mode of regulated cell death characterized by an iron-dependent form of phospholipid peroxidation induced by reactive oxygen species (ROS) [3, 4]. With the recognition that ferroptosis can be exploited to eradicate chemoresistance malignancies in an apoptosis-independent manner, several druggable targets for inducing ferroptosis have been discovered. These include: 1) direct inhibition of glutathione (GSH) production via class I ferroptosis inducers (FINs) (e.g., erastin [5], 5-octyl d-glutamate [6], sorafenib [7], sulfasalazine [8], acetaminophen [9], buthionine sulfoximine (BSO) [4]; 2) direct GPX4 inactivation using class II FINs (e.g., altretamine [10], ML-162 [4, 10, 11], ML-210 [4, 10, 11], RSL3 [3, 4], withaferin A [12], FIN56 [13, 14]), and 3) other noncanonical approaches such as iron-based nanoparticles and iron oxidizers (e.g., C′ dots [15], artemisinin [16], artesunate [17], FINO2 [14–17]), as well as some specific polyunsaturated fatty acids (PUFAs) and monounsaturated fatty acids (MUFAs) may provide alternative strategies to induce ferroptosis in cancers.
Ferroptosis execution can be categorized into canonical and noncanonical pathways [18], which refers to the inactivation of the central antioxidant system (canonical) and Fe2+ catalyzed ROS overload, respectively. However, distinct ferroptosis sensitivity patterns have been observed among various cancer cell types due to irregular iron metabolism [19], anti-oxidative defenses [20–22], and altered lipid metabolism [23]. Recently, the underlying mechanisms associated with ferroptosis susceptibility have been progressively revealed, especially in light of how cancer cells evolve adaptations to ferroptosis-resistant states in response to ferroptotic stress upon pharmacological interventions.
In accordance with the current molecular understanding of ferroptosis susceptibility for cancer treatment, this opinion article briefly summarizes the current understanding of ferroptosis as a means of chemoresistant cancer treatment. In particular, we highlight the role of iron and lipid metabolism in anti-oxidative defense. We also discuss emerging mechanisms regarding ferroptosis resistance to provide further insight for proposing new opportunities and potential pitfalls.
Programed cell death: the significance of ferroptosis
The phenomenon of ferroptosis in cancer cells originated in 2003 with the recognition that a specific quinazolinone compound erastin, promotes a novel iron-dependent cell death with the mutant RAS oncogene [24]. Later, this finding was further confirmed by identifying the Ras-selective lethal (RSL) small molecule RSL-3 and RSL-5 in 2008 [25]. Unlike other classic cell death modalities such as apoptosis, necroptosis, and autophagy, ferroptosis lacks nuclear morphological changes, cellular blebbing, and biochemical features involving caspase activation, LC3 conversion (LC3-I to LC3-II), and ATP depletion [3]. In contrast, this new mode of regulated cell death is characterized by morphological shrinkage and increased density of the mitochondrial membrane. At the biochemical level, ferroptosis mainly occurs with malfunctions of the system XC−/glutathione (GSH)/glutathione peroxidase 4 (GPX4) pathway and leads to uncontrolled peroxidation of phospholipids containing PFUs (PUFA-PLs) within the cell membranes [3, 4, 26]. The mechanistic relationship between lipid peroxidation and cell ferroptosis is still largely elusive. Recent studies have suggested that ROS free radicals could oxidize PUFA-PLs in cell membranes, and this consequence of the lipid compositional changes leads to membrane destruction, to membrane pores opening, and subsequent cell swelling followed by membrane rupture [27, 28]. Besides, this ferroptosis-associated cell rupture effect can spontaneously spread to neighboring cell populations through lipid peroxide in an iron-dependent manner [27]. Additionally, oxidized PUFAs and toxic by-products such as aldehydes and Michael acceptors may also disrupt the function and activity carried out by intracellular proteins and activate other fatal events downstream [29–31].
The role of iron metabolism in lipid peroxidation
Ferroptosis is an iron- and PUFA-PL peroxidation-dependent membrane dysfunction that results in regulated cell death [29]. The regulation of ferroptosis involves multiple genes and pathways associating with cellular homeostasis in iron metabolism, lipid synthesis, and redox status [18, 32]. PUFA-PLs were critical initiators of ferroptosis due to their high oxidation susceptibility, and this oxidative lipid damage can be driven by: 1) a free-radical chain reaction (autoxidation) and 2) an iron-dependent enzymatic oxygenation [33–35]. The initial step of the free-radical chain reaction is triggered by an ROS, including hydroxyl radicals (•OH) and lipid alkoxyl radicals generated from bio-Fenton and Haber–Weiss reactions (Equations 1 and 2), which require hydrogen peroxide (H2O2) and transition metals (presumably redox-active iron, Fe2+) as primary sources. Iron-dependent enzymatic lipid peroxidation is processed in a controlled manner via the lipoxygenase (LOX) family and cytochrome P450 oxidoreductase (POR) [26, 33].
Fenton (1) and Haber–Weiss (2) Reaction:
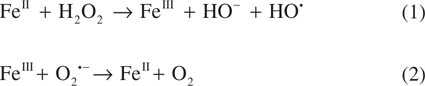
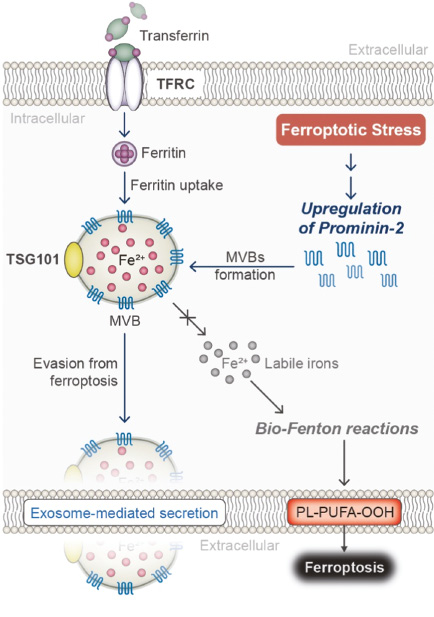
Free Fe2+ can be uptaken via the transferrin receptor (TFRC) and stored in ferritin cages, which undergoes ferritinophagy-mediated lysosomal degradation and releases labile iron to promote ferroptosis (Figure 1). Besides, iron sources are also stored intracellularly as heme and iron-sulfur clusters (ISCs). The cysteine desulfurase nitrogen fixation 1 (NFS1) that participates in ISC synthesis was found to be associated with ferroptosis susceptibility. The activation of NFS1 in cancer cells leads to a reduced level of labile irons, thereby losing the ferroptosis sensitivity [36]. CDGSH iron-sulfur domain-containing protein 1 (CISD-1) and CDGSH iron-sulfur domain-containing protein 2 (CISD-2) regulate the intracellular iron homeostasis; the overexpression of CISD-1 and CISD-2 can result in ferroptosis resistance against class I FINs [37, 38]. Recent studies reported that cancer cells could develop a natural protective mechanism in response to ferroptotic stress, including GPX4 inhibition and cell detachment from the extracellular matrix (ECM) (Figure 2). Upon the RNA-Seq screening of protein expression induced by pro-ferroptotic stimuli, prominin-2, a pentaspanin protein, was identified to be positively correlated with cell resistance to ferroptosis [19]. The key finding is that prominin-2 expression promoted the formation of ferritin-containing multivesicular bodies (MVBs) and exosomes that eliminate iron sources out of cells [19]. It was determined that the blockage of the prominin-2-mediated iron export pathway could restore the ferroptosis sensitivity to GPX4 inhibitors.
Figure 1 Several intracellular regulatory pathways of ferroptosis. TFRC: transferrin receptor; STEAP3: a six-transmembrane epithelial antigen of prostate 3; DMT1: divalent metal transporter 1; PUFA-PLs: phospholipids contain polyunsaturated fatty acids; LOX: lipoxygenase; POR: cytochrome P450 oxidoreductase; GSH: glutathione; GSSG: glutathione disulfide; GSR: glutathione S-reductase; GPX4: glutathione peroxidase 4; GLS: glutaminase; ACSL3: acyl-coenzyme A synthetase long-chain family member 3; MUFA: monounsaturated fatty acids; GCH1: GTP cyclohydrolase 1; BH4: tetrahydrobiopterin; MDM2: mouse double minute 2; MDMX: mouse double minute 4; PPARα: peroxisome proliferator-activated receptor-alpha; FSP1: ferroptosis suppressor protein 1; HMG-CoA: 3-hydroxy-3-methylglutaryl CoA; IPP: isopentenyl pyrophosphate; FPP: farnesyl pyrophosphate; CoQ10: coenzyme Q10.
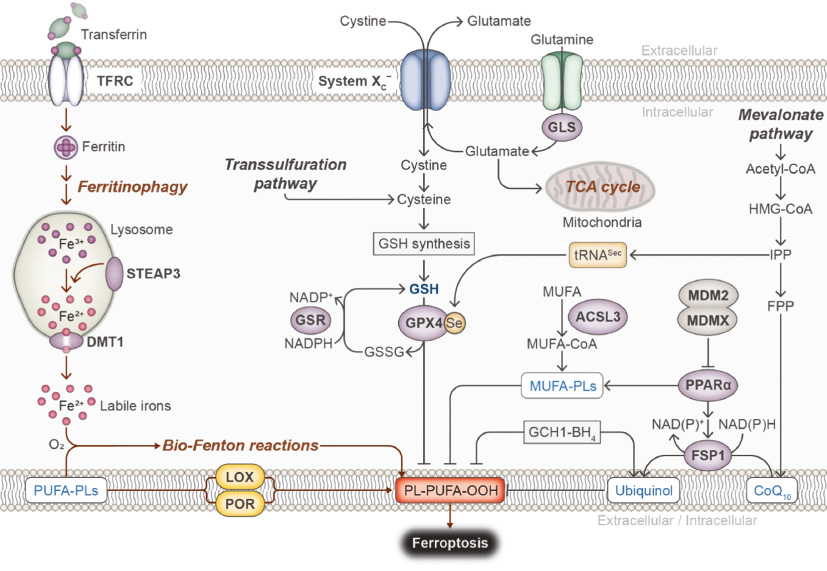
Figure 2 Schematic of the prominin-2-mediated exosomal release of iron, an emerging mechanism of ferroptosis escape. TSG101: tumor susceptibility 101; TFRC: transferrin receptor; MVBs: multivesicular bodies.
The anti-oxidative pathways regulate ferroptosis susceptibility
Lipid peroxidation and ferroptosis induced by ROS have been explored to address the chemoresistance of cancer cells [18, 39]. Considering the fact that cancer cells often suffer from a variable degree of oxidative stress during the tumor progression and therapeutic stimuli, but maintain at low lipid peroxidation rates, indicating their capacity to develop defensive antioxidant mechanisms [4, 19, 20, 23, 40, 41]. To date, some significant pathways have been identified that participate in maintaining oxidative homeostasis of membrane lipids and that can negatively regulate ferroptosis sensitivity, including: 1) the system XC−/GSH/GPX4 axis; 2) the Mevalonate-ferroptosis suppressor protein 1 (MVA-FSP1) axis; 3) the GTP cyclohydrolase 1-Tetrahydrobiopterin (GCH1-BH4) axis; 4) the peroxisome-ether-phospholipid axis; 5) acyl-CoA synthetase long-chain family member 3-monounsaturated fatty acids (ACSL3-MUFA) axis; and 6) the Liver kinase B1-AMP-activated protein kinase (LKB1-AMPK) axis. Therefore, current findings should collectively catalyze further studies to support the development of novel therapeutics against ferroptosis resistance.
GPX4 is a crucial glutathione peroxidase found in cellular membranes that repair preoxidized PL-PUFAs (Figure 1). In this regard, intracellular GSH is an antioxidant tripeptide that works as an essential cofactor to reduce lipid hydroperoxides [4]. De novo GSH synthesis requires a constant cysteine supply, which can either be produced from the transsulfuration pathway or imported from extracellular cystine via system XC− in exchange for intracellular glutamate [42]. Interference of system XC− activity through class I FINs (erastin, erastin2, 5-octyl D-glutamate, sorafenib, and sulfasalazine) blocks cystine uptake, resulting in insufficient GSH production [5]. Moreover, GSH-depleting agents (class I FINs) such as acetaminophen, n-acetyl-4-benzoquinone imine (NAPQI), and buthionine sulfoximine (BSO) can also lead to oxidative stress-induced ferroptosis [4, 9]. Alternatively, ferroptosis can be induced by direct inhibition of phospholipid peroxidase activity of GPX4 through covalent binding with the selenocysteine active site; these are known as class II FINs (e.g., altretamine, ML-162, ML-210, RSL3, and withaferin A) [10, 12, 26].
However, some tumor cells develop protective mechanisms enabling evasion from ferroptosis. The nuclear factor erythroid 2-related factor 2 (NRF2) is a key regulator in tumor cells that controls the expression of antioxidant proteins in response to ferroptosis stress [20]. Activation of the NRF2 pathway confers ferroptosis resistance to class I/II FINs. The inhibition/knockdown of NRF2 in hepatocellular carcinoma can increase sensitivity to erastin, sorafenib, and BSO [43]. Similarly, the acquired resistance to artesunate in head and neck cancer (HNC) cells could be reversed by NRF2 inhibition [21]. Moreover, inhibiting the NRF2 pathway in cisplatin-resistant cells (HN3R) and RSL3-resistant (HN3-rslR) cells yields enhanced sensitivity to RSL3 [22]. Thus, NRF2 could be a potent target for sensitizing class I/II FINs-resistant tumor cells.
In addition to the system XC−/GSH/GPX4 axis, the MVA-FSP1 axis is also crucial for GPX4 maturation and biosynthesis of coenzyme Q10 (CoQ10). GPX4 is a selenoprotein that requires the incorporation of selenocysteine (Sec) into its active site via the isopentenylation of tRNASec uses isopentenyl pyrophosphate (IPP), a central intermediate in the MVA pathway (Figure 1) [44]. Besides, the 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase is a rate-controlling enzyme of the MVA pathway (Figure 1). Thus, HMG-CoA reductase inhibitors, often known as statins, act by blocking the GPX4 and CoQ10 biosynthesis via inhibition of IPP formation [45]. Like the oxime-containing molecule FIN56, several compounds promote the degradation of GPX4 and interfere with the CoQ10 biosynthesis [13]. Reduced CoQ10 (ubiquinol) is an endogenous antioxidant produced by the MVA-FSP1 pathway [46]. Of note, the antioxidant capacity of ubiquinol is maintained by membrane-tethered ferroptosis- suppressor protein 1 (FSP1), an NADH-dependent CoQ oxidoreductase that reduces CoQ10 to ubiquinol. CoQ10 synthesis and FSP1 machinery given their central role in the MVA-FSP1 axis, which has been validated as a second antioxidant system that acts fully independent of the XC−/GSH/GPX4 axis [41, 47], suggests that FSP1 is a vital ferroptosis resistance factor that can be targeted to sensitize specific ferroptosis-resistant cancer cells [41]. FSP1 can be activated by a nuclear receptor protein, PPARa, dependent on upstream negative regulatory MDM2/MDMX complex, in a p53-independent manner (Figure 1) [48].
In a related vein, the ubiquinol level can also be reinforced by GTP cyclohydrolase-1 (GCH1) generated tetrahydrobiopterin (BH4) upon ferroptosis-induction (Figure 1) [49, 50]. Moreover, the production of BH4 enables lipid remodeling with its direct antioxidant effect to protect specific PUFA-PLs from oxidative degradation [49, 50]. Therefore, the GCH1-BH4 axis could be considered a potential target for those cancer cells resistant to GPX4 inhibition.
With respect to the lipid metabolism associated with ferroptosis susceptibility, it was determined that the polyunsaturated ether phospholipids (PUFA-ePLs) act as a key pro-ferroptotic substrate for lipid peroxidation and is closely associated with ferroptosis execution [23]. The biosynthesis of PUFA-ePLs is driven by peroxisome biogenesis [51, 52], which sufficiently controls sensitivity to ferroptosis (Figure 3) [23]. In this sense, the peroxisome-ether-phospholipid axis has been proposed to explain how adaptive evasion of ferroptosis induction can occur in some carcinoma cells. The study has uncovered that the initial ferroptosis-sensitive cells could adapt to survive under oxidative stress through significant downregulation of PUFA-ePLs [23]. This suggests that the modulation of peroxisome and PUFA-ePLs levels could help promote the ferroptotic process in cancer treatment.
Figure 3 Schematic of the emerging mechanisms of underlying susceptibility and resistance to ferroptosis. Lipid metabolic dynamics and membrane lipid composition are closely connected to the ferroptosis-resistant cell state. ER: endoplasmic reticulum; DGLA: dihomo-γ-linolenic acid; LKB1-AMPK: serine-threonine kinase liver kinase B1-AMP-activated protein kinase; ACC1: acetyl-CoA carboxylase 1; PUFA: polyunsaturated fatty acids.
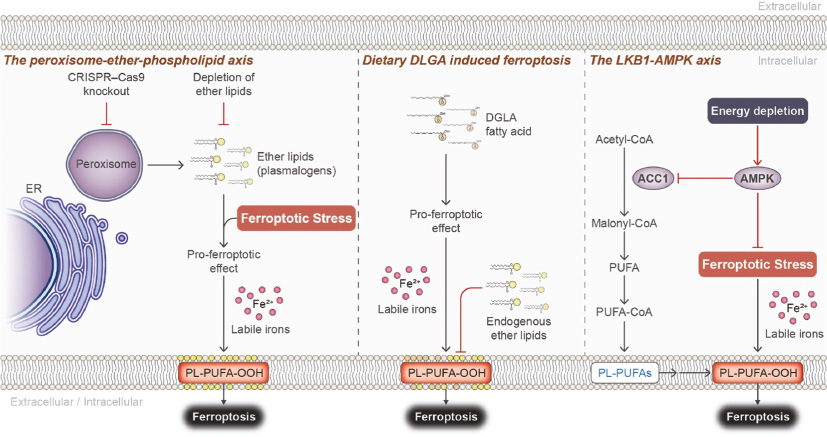
Besides, a recent study has explored a new potent ferroptosis inducer dihomo-γ-linolenic acid (DGLA), an exogenous PUFAs that appears to have a unique pro-ferroptotic effect distinct from other exogenous PUFAs, including arachidonic acid, eicosapentaenoic acid, and docosahexaenoic acid, which did not induce Fer-1-sensitive ferroptosis alone [53]. The mechanism of DGLA-induced ferroptosis was proposed through the direct modulation of membrane phospholipid acylation (Figure 3) [53]. Notably, the evidence indicates that endogenous ether lipids (mainly plasmalogens) could act as antioxidant molecules to protect both Caenorhabditis elegans and cancer cells from ferroptosis induced by exogenous DGLA [53, 54]. These findings imply that endogenous biosynthesis of plasmalogens can play distinct roles in response to specific ferroptotic stimuli. Moreover, a ferroptosis-resistant cell state can also be triggered by exogenous and endogenous MUFAs [29, 53, 55]. Acyl-CoA synthetase long-chain family member 3 (ACSL3) is required for MUFA activation. Once activated, MUFAs can incorporate into the plasma membrane and reduce the ferroptotic sensitivity by altering PUFAs composition of the plasma membrane [55].
Interestingly, recent studies found that ATP depletion in cancer cells can activate AMP-activated protein kinase (AMPK), a ubiquitous cellular energy sensor that confers resistance to ferroptosis by negatively regulates acetyl-CoA carboxylase (ACC) and PUFA biosynthesis (Figure 3) [56–58]. It has been confirmed that a key serine-threonine kinase encoded by LKB1 activates the AMPK through direct phosphorylation, which is one of the frequently mutated tumor suppressors in several human cancers [59]. Further investigation indicated that LKB1-AMPK negatively regulates the rate-limiting enzyme ACC1 during the PUFA biosynthesis upon the energy stress [57]. Collectively, the study concluded that LKB1-AMPK-dependent ACC1-mediated fatty acid biogenesis is essential for ferroptosis execution. In line with these findings, gaining a more in-depth understanding of the regulation in lipid metabolic dynamics and membrane lipid composition is essential for developing novel strategies against ferroptosis resistance.
Perspectives
The discovery of ferroptosis offered a tremendous therapeutic opportunity in cancer treatment, and its practical effectiveness in clinical settings is steadily progressing. Several Food and Drug Administration-approved drugs, including altretamine, sorafenib, artesunate, and artemisinin, have been recognized with pro-ferroptotic effect. A number of FINs have been developed and verified in preclinical cancer models [60]. However, recent studies have identified new metabolic pathways that confer resistance to cancer cell ferroptosis induction, particularly the role of lipid metabolism in the regulation of ferroptosis sensitivity [19, 23, 53, 57]. A deeper understanding of mechanisms driving ferroptosis-sensitive state remains further investigation. In this regard, targeting three hallmark features (i.e., PUFA-PLs, redox-active iron, and antioxidant system) of ferroptosis provides effective routes to eradicating aggressive malignancies. Aside from ferroptosis mechanistic studies, interdisciplinary approaches with the integration of nanomedicine, immunotherapy, and radiotherapy hold additional opportunities to promote ferroptosis-based therapeutic approaches toward clinical translation.
Acknowledgments
This work is supported in part by the National Natural Science Foundation of China (J.W., 82001887and Z.W. 81900105). J.W. was supported by a grant from the Hundred Talents Program (75110-18841227) from Sun Yat-sen University and the Guangdong Basic and Applied Basic Research Foundation (2019A1515110326).
References
- Su Z, Yang Z, Xie L, DeWitt JP, Chen Y. Cancer therapy in the necroptosis era. Cell Death Differ 2016;23:748-56. [PMID: 26915291 DOI: 10.1186/s12943-019-1029-8]
- Holohan C, Van Schaeybroeck S, Longley DB, Johnston PG. Cancer drug resistance: an evolving paradigm. Nat Rev Cancer 2013;13:714-26. [PMID: 24060863 DOI: 10.1038/nrc3599]
- Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 2012;149:1060-72. [PMID: 22632970 DOI: 10.1016/j.cell.2012.03.042]
- Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, et al. Regulation of ferroptotic cancer cell death by GPX4. Cell 2014;156:317-31. [PMID: 24439385 DOI: 10.1016/j.cell.2013.12.010]
- Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, et al. Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 2014;3:e02523. [PMID: 24844246 DOI: 10.7554/eLife.02523.001]
- Chin RM, Fu X, Pai MY, Vergnes L, Hwang H, et al. The metabolite alpha-ketoglutarate extends lifespan by inhibiting ATP synthase and TOR. Nature 2014;510:397–401. [PMID: 24828042 DOI: 10.1038/nature13264]
- Louandre C, Ezzoukhry Z, Godin C, Barbare JC, Maziere JC, et al. Iron-dependent cell death of hepatocellular carcinoma cells exposed to sorafenib. Int J Cancer 2013;133:1732-42. [PMID: 23505071 DOI: 10.1002/ijc.28159]
- Gout PW, Buckley AR, Simms CR, Bruchovsky N. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)-cystine transporter: a new action for an old drug. Leukemia 2001;15:1633-40. [PMID: 11587223 DOI: 10.1038/sj.leu.2402238]
- Lorincz T, Jemnitz K, Kardon T, Mandl J, Szarka A. Ferroptosis is involved in acetaminophen induced cell death. Pathol Oncol Res 2015;21:1115-21. [PMID: 25962350 DOI: 10.1007/s12253-015-9946-3]
- Woo JH, Shimoni Y, Yang WS, Subramaniam P, Iyer A, et al. Elucidating compound mechanism of action by network perturbation analysis. Cell 2015;162:441-51. [PMID: 26186195 DOI: 10.1016/j.cell.2015.05.056]
- Weiwer M, Bittker JA, Lewis TA, Shimada K, Yang WS, et al. Development of small-molecule probes that selectively kill cells induced to express mutant RAS. Bioorg Med Chem Lett 2012;22:1822-6. [PMID: 22297109 DOI: 10.1016/j.bmcl.2011.09.047]
- Hassannia B, Wiernicki B, Ingold I, Qu F, Van Herck S, et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. J Clin Invest 2018;128:3341-55. [PMID: 29939160 DOI: 10.1172/JCI99032]
- Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, et al. Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 2016;12:497-503. [PMID: 27159577 DOI: 10.1038/nchembio.2079]
- Gaschler MM, Andia AA, Liu H, Csuka JM, Hurlocker B, et al. FINO2 initiates ferroptosis through GPX4 inactivation and iron oxidation. Nat Chem Biol 2018;14:507-15. [PMID: 29610484 DOI: 10.1038/s41589-018-0031-6]
- Kim SE, Zhang L, Ma K, Riegman M, Chen F, et al. Ultrasmall nanoparticles induce ferroptosis in nutrient-deprived cancer cells and suppress tumour growth. Nat Nanotechnol 201;11:977-85. [PMID: 27668796 DOI: 10.1038/nnano.2016.164]
- Chen GQ, Benthani FA, Wu J, Liang D, Bian ZX, et al. Artemisinin compounds sensitize cancer cells to ferroptosis by regulating iron homeostasis. Cell Death Differ 2020;27:242-54. [PMID: 31114026 DOI: 10.1038/s41418-019-0352-3]
- Eling N, Reuter L, Hazin J, Hamacher-Brady A, Brady NR. Identification of artesunate as a specific activator of ferroptosis in pancreatic cancer cells. Oncoscience 2015;2:517-32. [PMID: 26097885 DOI: 10.18632/oncoscience.160]
- Hassannia B, Vandenabeele P, Vanden Berghe T. Targeting ferroptosis to iron out cancer. Cancer Cell 2019;35:830-49. [PMID: 31105042 DOI: 10.1016/j.ccell.2019.04.002]
- Brown CW, Amante JJ, Chhoy P, Elaimy AL, Liu H, et al. Prominin2 drives ferroptosis resistance by stimulating iron export. Dev Cell 2019;51:575-86 e4. [PMID: 31735663 DOI: 10.1016/j.devcel.2019.10.007]
- Dodson M, Castro-Portuguez R, Zhang DD. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biol 2019;23:101107. [PMID: 30692038 DOI: 10.1016/j.redox.2019.101107]
- Roh JL, Kim EH, Jang H, Shin D. Nrf2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biol 2017;11:254-62. [PMID: 28012440 DOI: 10.1016/j.redox.2016.12.010]
- Shin D, Kim EH, Lee J, Roh JL. Nrf2 inhibition reverses resistance to GPX4 inhibitor-induced ferroptosis in head and neck cancer. Free Radic Biol Med 2018;129:454-62. [PMID: 30339884 DOI: 10.1016/j.freeradbiomed.2018.10.426]
- Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, et al. Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 2020;585:603-8. [PMID: 32939090 DOI: 10.1038/s41586-020-2732-8]
- Dolma S, Lessnick SL, Hahn WC, Stockwell BR. Identification of genotype-selective antitumor agents using synthetic lethal chemical screening in engineered human tumor cells. Cancer Cell 2003;3:285-96. [PMID: 12676586 DOI: 10.1016/s1535-6108(03)00050-3]
- Yang WS, Stockwell BR. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 2008;15:234-45. [PMID: 18355723 DOI: 10.1016/j.chembiol.2008.02.010]
- Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, et al. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 2016;113:E4966-75. [PMID: 27506793 DOI: 10.1073/pnas.1603244113]
- Riegman M, Sagie L, Galed C, Levin T, Steinberg N, et al. Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol 2020;22:1042-8. [PMID: 32868903 DOI: 10.1038/s41556-020-0565-1]
- Agmon E, Solon J, Bassereau P, Stockwell BR. Modeling the effects of lipid peroxidation during ferroptosis on membrane properties. Sci Rep 2018;8:5155. [PMID: 29581451 DOI: 10.1038/s41598-018-23408-0]
- Stockwell BR, Friedmann Angeli JP, Bayir H, Bush AI, Conrad M, et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell 2017;171:273-85. [PMID: 28985560 DOI: 10.1016/j.cell.2017.09.021]
- Feng H, Stockwell BR. Unsolved mysteries: how does lipid peroxidation cause ferroptosis? PLoS Biol 2018;16:e2006203. [PMID: 29795546 DOI: 10.1371/journal.pbio.2006203]
- Magtanong L, Ko PJ, Dixon SJ. Emerging roles for lipids in nonapoptotic cell death. Cell Death Differ 2016;23:1099-109. [PMID: 26967968 DOI: 10.1038/cdd.2016.25]
- Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ 2018;25:486-541. [PMID: 29362479 DOI: 10.1038/s41418-017-0012-4]
- Zou Y, Li H, Graham ET, Deik AA, Eaton JK, et al. Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 2020;16:302-9. [PMID: 32080622 DOI: 10.1038/s41589-020-0472-6]
- Kagan VE, Mao G, Qu F, Angeli JP, Doll S, et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 2017;13:81–90. [PMID: 27842066 DOI: 10.1038/nchembio.2238]
- Conrad M, Pratt DA. The chemical basis of ferroptosis. Nat Chem Biol 2019;15:1137-47. [PMID: 31740834 DOI: 10.1038/s41589-019-0408-1]
- Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, et al. NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 2017;551:639-43. [PMID: 29168506 DOI: 10.1038/nature24637]
- Kim EH, Shin D, Lee J, Jung AR, Roh JL. CISD2 inhibition overcomes resistance to sulfasalazine-induced ferroptotic cell death in head and neck cancer. Cancer Lett 2018;432:180-90. [PMID: 29928961 DOI: 10.1016/j.canlet.2018.06.018]
- Yuan H, Li X, Zhang X, Kang R, Tang D. CISD1 inhibits ferroptosis by protection against mitochondrial lipid peroxidation. Biochem Biophys Res Commun 2016;478:838-44. [PMID: 27510639 DOI: 10.1016/j.bbrc.2016.08.034]
- Friedmann Angeli JP, Krysko DV, Conrad M. Ferroptosis at the crossroads of cancer-acquired drug resistance and immune evasion. Nat Rev Cancer 2019;19:405-14. [PMID: 31101865 DOI: 10.1038/s41568-019-0149-1]
- Li D, Li Y. The interaction between ferroptosis and lipid metabolism in cancer. Signal Transduct Target Ther 2020;5:108. [PMID: 32606298 DOI: 10.1038/s41392-020-00216-5]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, et al. FSP1 is a glutathione-independent ferroptosis suppressor. Nature 2019;575:693-8. [PMID: 31634899 DOI: 10.1038/s41586-019-1707-0]
- Sato H, Tamba M, Ishii T, Bannai S. Cloning and expression of a plasma membrane cystine/glutamate exchange transporter composed of two distinct proteins. J Biol Chem 1999;274:11455-8. [PMID: 10206947 DOI: 10.1074/jbc.274.17.11455]
- Sun X, Ou Z, Chen R, Niu X, Chen D, et al. Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 2016;63:173-84. [PMID: 26403645 DOI: 10.1002/hep.28251]
- Warner GJ, Berry MJ, Moustafa ME, Carlson BA, Hatfield DL, et al. Inhibition of selenoprotein synthesis by selenocysteine tRNA[Ser]Sec lacking isopentenyladenosine. J Biol Chem 2000;275:28110-9. [PMID: 10821829 DOI: 10.1074/jbc.M001280200]
- Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, et al. Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 2017;551:247-50. [PMID: 29088702 DOI: 10.1038/nature24297]
- Hadian K. Ferroptosis suppressor protein 1 (FSP1) and coenzyme Q10 cooperatively suppress ferroptosis. Biochemistry 2020;59:637-8. [PMID: 32003211 DOI: 10.1021/acs.biochem.0c00030]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, et al. The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 2019;575:688-92. [PMID: 31634900 DOI: 10.1038/s41586-019-1705-2]
- Venkatesh D, O’Brien NA, Zandkarimi F, Tong DR, Stokes ME, et al. MDM2 and MDMX promote ferroptosis by PPARalpha-mediated lipid remodeling. Genes Dev 2020;34:526-43. [PMID: 32079652 DOI: 10.1101/gad.334219.119]
- Kraft VAN, Bezjian CT, Pfeiffer S, Ringelstetter L, Muller C, et al. GTP Cyclohydrolase 1/tetrahydrobiopterin counteract ferroptosis through lipid remodeling. ACS Cent Sci 2020;6:41-53. [PMID: 31989025 DOI: 10.1021/acscentsci.9b01063]
- Soula M, Weber RA, Zilka O, Alwaseem H, La K, et al. Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 2020;16:1351-60. [PMID: 32778843 DOI: 10.1038/s41589-020-0613-y]
- Lodhi IJ, Semenkovich CF. Peroxisomes: a nexus for lipid metabolism and cellular signaling. Cell Metab 2014;19:380-92. [PMID: 24508507 DOI: 10.1016/j.cmet.2014.01.002]
- Honsho M, Fujiki Y. Plasmalogen homeostasis – regulation of plasmalogen biosynthesis and its physiological consequence in mammals. FEBS Lett 2017;591:2720-9. [PMID: 28686302 DOI: 10.1002/1873-3468.12743]
- Perez MA, Magtanong L, Dixon SJ, Watts JL. Dietary lipids induce ferroptosis in Caenorhabditis elegans and human cancer cells. Dev Cell 2020;54:447-54.e4. [PMID: 32652074 DOI: 10.1016/j.devcel.2020.06.019]
- Shi X, Tarazona P, Brock TJ, Browse J, Feussner I, et al. A Caenorhabditis elegans model for ether lipid biosynthesis and function. J Lipid Res 2016;57:265-75. [PMID: 26685325 DOI: 10.1194/jlr.M064808]
- Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, et al. Exogenous monounsaturated fatty acids promote a ferroptosis-resistant cell state. Cell Chem Biol 2019;26:420-32.e9. [PMID: 30686757 DOI: 10.1016/j.chembiol.2018.11.016]
- Lee H, Zandkarimi F, Zhang Y, Meena JK, Kim J, et al. Energy-stress-mediated AMPK activation inhibits ferroptosis. Nat Cell Biol 2020;22:225-34. [PMID: 32029897 DOI: 10.1038/s41556-020-0461-8]
- Li C, Dong X, Du W, Shi X, Chen K, et al. LKB1-AMPK axis negatively regulates ferroptosis by inhibiting fatty acid synthesis. Signal Transduct Target Ther 2020;5:187. [PMID: 32883948 DOI: 10.1038/s41392-020-00297-2]
- Hardie DG, Ross FA, Hawley SA. AMPK: a nutrient and energy sensor that maintains energy homeostasis. Nat Rev Mol Cell Biol 2012;13:251-62. [PMID: 22436748 DOI: 10.1038/nrm3311]
- Herzig S, Shaw RJ. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat Rev Mol Cell Biol 2018;19:121-35. [PMID: 28974774 DOI: 10.1038/nrm.2017.95]
- Liang C, Zhang X, Yang M, Dong X. Recent progress in ferroptosis inducers for cancer therapy. Adv Mater 2019;31:e1904197. [PMID: 31595562 DOI: 10.1002/adma.201904197]