A Review for Antimicrobial Peptides with Anticancer Properties: Re-purposing of Potential Anticancer Agents
1Breast Tumor Center, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou 510120, China
2Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou 510120, China
3Guangzhou Regenerative Medicine and Health Guangdong Laboratory, Guangzhou 510005, China
4CAS Key Laboratory of Quantitative Engineering Biology, Shenzhen Institute of Synthetic Biology, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen, Guangdong 518055, China
5Department of Clinical Laboratory, Sun Yat-sen Memorial Hospital, Sun Yat-sen University, Guangzhou 510120, China
*Correspondence to: Songyin Huang E-mail: huangsy@mail.sysu.edu.cn Yandan Yao E-mail: yaoyand@mail.sysu.edu.cn
Received: June 14 2020; Revised: August 17 2020; Accepted: August 18 2020; Published Online: November 18 2020
Cite this paper:
Cuiyu Zhong, Lei Zhang, Lin Yu, Jiandong Huang, Songyin Huang and Yandan Yao. A Review for Antimicrobial Peptides with Anticancer Properties: Re-purposing of Potential Anticancer Agents. BIO Integration 2020; 1(4): 156–167.
DOI: 10.15212/bioi-2020-0013. Available at: https://bio-integration.org/
Download citation 
© 2020 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
In recent years, various research on cancer treatment has achieved significant progress. However, some of these treatments remain disputable because of the emergence and development of drug resistance, and the toxic side effects that were brought about by the lack of selectivity displayed by the treatments. Hence, there is considerable interest in a new class of anticancer molecules that is currently still under investigation termed the cationic antimicrobial peptides (AMPs). AMPs are a group of pervasive components of the innate immunity which can be found throughout all classes of life. The small innate peptides cover a broad spectrum of antibacterial activities due to their electrostatic interactions with the negatively charged bacterial membrane. Compared with normal cells, cancer cells have increased proportions of negatively charged molecules, including phosphatidylserine, glycoproteins, and glycolipids, on the outer plasma membrane. This provides an opportunity for exploiting the interaction between AMPs and negatively charged cell membranes in developing unconventional anticancer strategies. Some AMPs may also be categorized into a group of potential anticancer agents called cationic anticancer peptides (ACPs) due to their relative selectivity in cell membrane penetration and lysis, which is similar to their interaction with bacterial membranes. Several examples of ACPs that are used in tumor therapy for their ability in penetrating or lysing tumor cell membrane will be reviewed in this paper, along with a discussion on the recent advances and challenges in the application of ACPs.
Keywords
Anticancer activity, anticancer peptide, antimicrobial peptide, electrostatic interaction, membrane, nanosystems.
Introduction
Despite the numerous breakthroughs in the field of medical research over the past few years, cancer remains one of the leading causes of death in humans [1–3]. At present, conventional chemotherapy is still the preferred method of cancer treatment, even though its effectiveness is often hindered by inherent or acquired drug resistance [4, 5]. There are many reasons for chemotherapeutic resistance at the tissue and cellular level. It can be caused by the metabolic heterogeneity of tumor cells, where cell populations with different, complementary metabolic profiles couple up to induce chemotherapeutic resistance and cancer progression [6, 7]. In addition, the activation of growth signaling pathways or the inhibition of apoptosis, upregulation of drug transporters, and deoxyribo nucleic acid (DNA) damage repair are among some of the other common mechanisms for the development of drug resistance in cancer cells [8–10].
Antimicrobial peptides (AMPs) are part of the innate immune system in many organisms, and their potential in combating bacterial infections has been studied in depth [11]. Lately, researchers have discovered a group of AMPs that are involved in both antimicrobial and anticancer activities, nomenclated as anticancer peptides (ACPs). The novel mechanism of ACPs leads to lower possibility in the development of resistance compared to conventional chemotherapy [12–14]. Some ACPs can be utilized as an efficient strategy for intracellular drug delivery in cancer treatment which greatly improves drug permeation and the therapeutic effect [15]. There are also studies that demonstrate the ability of ACPs to lyse tumor cells directly [16, 17]. However, the application of ACPs in antitumor therapy is still primitive and requires further research. In this paper, the potential of ACPs in antitumor therapy through penetrating or lysing membranes is reviewed. A brief overview is also presented on other antitumor mechanisms of ACPs, such as apoptosis induction, anti-angiogenesis, immune cells recruitment, and so on [18, 19].
Membrane interaction mechanism of ACPs
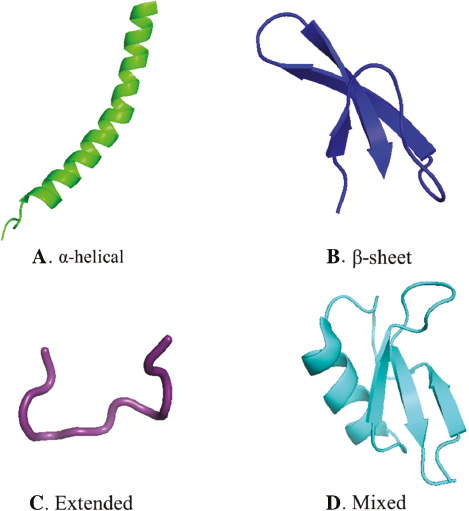
Most ACPs are short peptides, approximately 50–60 amino acid residues in length [20, 21]. Despite their diversity in size and shape, be it as primary or secondary structures (such as α-helix, β-sheets, extended helix, and mixed as shown in Figure 1), most AMPs are cationic, amphipathic and possess a signi?cant proportion of hydrophobic residues [22]. The cationic surface charges of ACPs are responsible for the electrostatic interactions between ACPs and negatively charged lipids on the cell membranes [23]. Meanwhile, the hydrophobic domain plays a vital role in the mechanism of the subsequent actions of ACPs after the initial interactions between ACPs and the cell membrane [24]. Yin et al. demonstrated that cationic surface charges of ACPs and an optimum hydrophobic domain are important factors affecting the function of ACPs, and the balance between the charge distribution and hydrophobicity of ACPs would promote the membrane-lysing activity [25]. These physicochemical properties lay the foundation for the activity of ACPs against bacteria and tumor.
Figure 1 Major structural classes of ACPs. The representative structures of four main classes are given. A. The human cathelicidin antimicrobial peptide, LL-37 is an α-helical [88] (PDB code: 2K6O). B. The human α-dedensin-1, HNP-1, contains a triple-stranded β-sheet [16] (PDB code: 2PM1). C. The bovine neutrophil peptide, indolicidin, does not contain α-helices or β-strands [89] (PDB code: 1QXQ). D. The ornamental tobacco defensin, NaD1, comprises a triple-stranded β-sheet with one α-helix [16] (PDB code: 1MR4). Images generated using PyMol (DeLano Scientific LLC, CA USA).
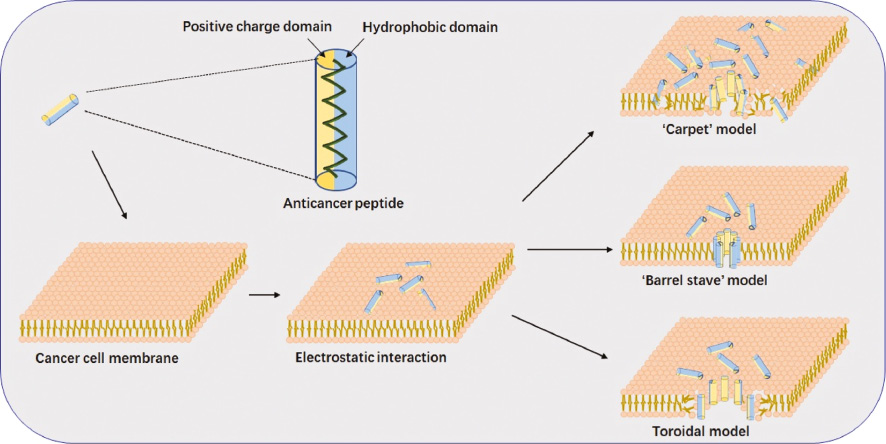
Several models have been reported to describe the various methods that ACPs utilize in inducing membrane destabilization and permeabilization or peptide internalization, among these are the carpeting, toroidal pore, and barrel-stave models (Figure 2) [26]. The carpet model describes a process of parallel peptide accumulation on the anionic cell surface via electrostatic interactions just like a carpet. Like detergents, the curvature strain of the membrane induces disruption on the membrane after peptides accumulate to a certain concentration. ACPs may then enter the cell membrane without the formation of a stable pore or causing membrane disintegration [27]. Järvå et al. proved that defensins, cationic antimicrobial peptides, disturb the cell membrane through the carpet model by observing a carpet-like antimicrobial defensin–phospholipid membrane disruption complex on X-ray and providing a high-resolution image of a carpet-like mechanism of action [28]. In the barrel-stave model, a large number of peptides interact with the cell membrane and accumulate on the membrane surface, causing conformational changes. After that, the hydrophobic amino acid of peptides will be exposed to the hydrophobic core of the lipid bilayer, subsequently causing the leakage of cell contents through the formation of transmembrane pores by ACPs [11]. Yang et al. demonstrated that alamethicin, a natural peptide, conformed to the barrel-stave model [29]. To some extent, the toroidal model is similar to the barrel-stave model, whereby both models ultimately lead to the leakage of cell contents because peptides can span the membrane to form a pore [30]. However, head groups of membrane lipids must remain in contact with the hydrophilic domain of the ACPs throughout the membrane to make ‘toroidal-like’ pores in the phospholipid layer of the toroidal model [31]. For example, LL-37 could disrupt the cell membrane through the toroidal pore mechanism, having been confirmed by Wildman et al. [32].
Figure 2 Membrane penetration or lysis mechanism of ACPs mainly include three modes: carpeting, toroidal pore, and barrel-stave models. In the carpet model, ACPs bind to phospholipid head groups via electrostatic interactions and align themselves parallel to the membrane surface in a carpet-like fashion until a critical threshold concentration is reached. Subsequently, ACPs may enter the cell membrane without the formation of a pore or causing membrane disruption. In the barrel-stave model, peptides self-aggregate in the membrane when a critical threshold concentration of peptide is reached, causing conformational changes. And then, the hydrophobic amino acid of peptides will be exposed to the hydrophobic core of the lipid bilayer and a transmembrane pore is formed of which the hydrophilic face forms the inner channel while the hydrophobic face is on the outside. The toroidal pore model is similar to the barrel-stave model except that the headgroups of membrane lipids must remain associated with the hydrophilic domain of the peptides throughout the membrane in this model to make toroidal-like pores in the phospholipid layer.
ACPs specifically target cancer cells because of their negatively charged membranes. Similar to bacteria, cancer cells have negatively charged membranes because of the overexpression of anionic molecules such as phospholipid phosphatidylserine (PS) [33], heparin sulfate, sialylated gangliosides, and O-glycosylated mucins on their membrane surface [34]. On the contrary, there are more zwitterionic phospholipids, including phosphatidylcholine (PC), phosphatidylethanolamine (PE), and sphingomyelin (SM), on the membrane surface of normal cells [35]. Leite et al. found that the enrichment of PS in the outer leaflet of cancer cells significantly promoted the binding of ACPs to cancer cell membranes [36]. Therefore, ACPs would interact with the membranes of cancer cells instead of normal cells. It should, however, be noted that the anticancer mechanism of ACPs is not only limited to membrane disturbance. Some ACPs can fight tumors via other mechanisms such as affecting immunity and intracellular killing pattern, which will be discussed in a later part of this paper [19, 37].
ACPs treat tumors by lysing or penetrating the membrane
Most ACPs can selectively attach to and lyse cancer cell membranes, consequently leading to the leakage of cellular contents and cell death (Figure 3) [35]. Due to its unique action mechanism, ACP works even before it gets into the cells, lowering the possibility of drug resistance as compared to conventional chemotherapeutic drugs [38]. At the same time, negative charges on cancer cell membranes allow a certain degree of selectivity by ACPs [22]. Studies have also shown that although many ACPs adopt an irregular structure in aqueous solution, they are converted into a bioactive, helical conformation at the negatively charged membrane [27, 30]. Most ACPs take the helical structure as the bioactive conformation, but there are a few exceptions that adopt the β-structure instead, which fold prior to engaging the membrane [39]. This characteristic also contributes to the selectivity of ACPs. Therefore, ACPs present a novel strategy in tumor treatment. In recent decades, many researchers have been working to exploit the potential of ACPs in cancer treatment by cell membrane lysis, where some of them are already at the stage of clinical trials [40]. There is a list of representative ACPs that demonstrates their anticancer activity against several cancer types (Table 1).
Figure 3 Different mechanisms of anticancer peptides. One of the main anticancer mechanisms of ACPs is membrane disturbance including lysing and penetrating the membrane. In addition, some ACPs can also fight against cancer through other mechanisms such as angiogenesis inhibition, tumor apoptosis induction, and essential cell protein targeting, or immune cell recruitment to attack cancer cells as described.
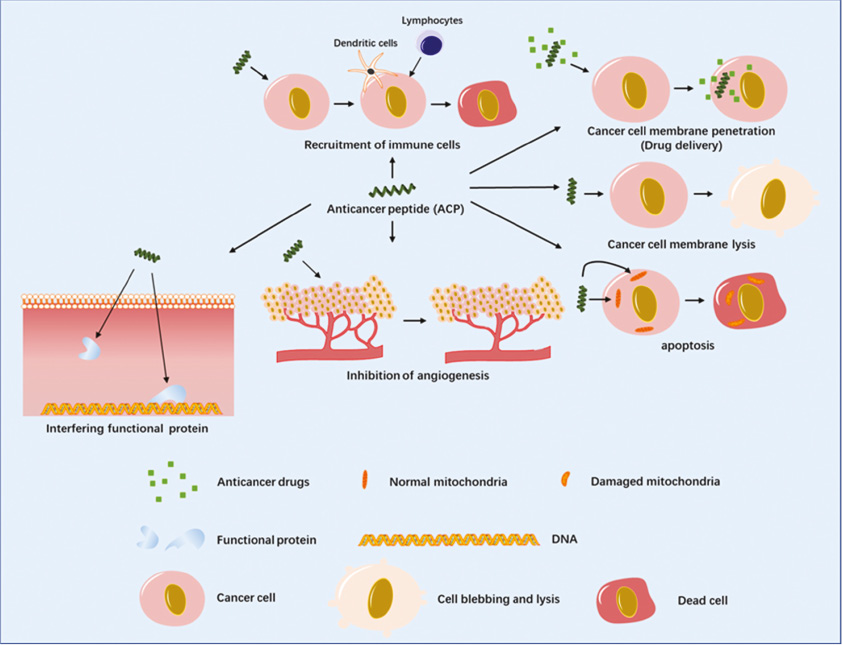
Table 1 Representative ACPs that Showed Anticancer Activity Against Several Cancer Types
| ACP | Source | Target Cancer | Anticancer activity | Ref. |
|---|---|---|---|---|
| Melittin | Apis mellifera | Human and murine leukemic cells | Membrane permeabilization; phospholipase A2 activator; Phospholipase D activator | [90–94] |
| Polybia-MP1 | Polybia paulista (Brazilian wasp) | Human prostate, bladder, and multi- resistant leukemic cancer cells | Membrane permeabilization | [36, 43, 95] |
| NRC-03, NRC-07 | Atlantic flounder species | Human breast cancer cells; murine mammary carcinoma cells | Membrane permeabilization | [53] |
| D-peptides A, B, C, and D | Synthetic | Human, lung, cervix, glioma cancer cell; mouse myeloma cells; African green monkey kidney cancer cells | Membrane permeabilization | [96] |
| Magainin 2 | Xenopus laevis | Human bladder cancer cells | Membrane permeabilization; apoptosis inducer? | [42, 97, 98] |
| Gomesin | Brazilian spider | Human colon, breast, and cervix adenocarcinoma cancer cells; murine melanoma | Membrane permeabilization | [54] |
| SVS-1 | Synthetic | Human epidermis, breast, and lung cancer cells | Membrane permeabilization | [39, 99] |
| Lactoferrici B | Bos taurus | Human fibrosarcoma, leukemia, various carcinomas, colon, gastric, neuroblastoma, ovarian, and breast carcinoma cells; murine melanoma and lymphoma cells | Membrane permeabilization; apoptosis inducer; antiangiogenic; late-stage inhibition of autophagy | [100–104] |
| LL-37 | Homo sapiens | Human oral squamous cell carcinoma and leukemic cells | Membrane permeabilization; caspase-independent apoptosis | [32, 105, 106] |
| LTX-315 | Synthetic | Human osteosarcoma cells; murine melanoma breast cancer cells; rat fibrosarcoma cells | Membrane permeabilization; Immunological cell death | [107–111] |
| Pardaxin | Fish | Human oral squamous cell carcinoma cells | Membrane permeabilization | [112] |
The outline of the mechanism in fighting cancer is similar among different ACPs, but there are some distinct differences among them when it comes to their specific effects. ACPs such as melittin can interact with cancer cell membranes within a short period of time and cause membrane destruction as cell as swelling, membrane blebbing, and breakage [37, 41]. The anticancer peptide magainin II, extracted from the skin of the African clawed frog, can selectively kill bladder cancer cells in vitro by the forming channels in cell membranes. Although this ultimately leads to cell death, it hardly affects normal cells at all [42]. Polybia-MPI, an antimicrobial peptide purified from the venom of the social wasp Polybia paulista, can selectively kill prostate and bladder cancer cells by membrane destruction, but barely affects normal murine fibroblasts [43].
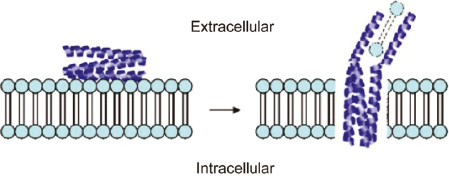
Wang et al. reported that polybia-MPI could cause necrosis of various leukemia cells through forming transmembrane pores or membrane destruction, acting at the cell membrane level instead of entering the cell (Figure 4). This makes polybia-MPI immune to the common mechanisms of multidrug resistance, eliminating the possibility of cancer cells developing resistance during treatment [38]. Sinthuvanich et al. designed an 18-residue peptide, SVS-1, which tends to fold at the surface of negatively charged cancer cell membranes, and adopts an amphiphilic β-hairpin structure capable of membrane destruction, but remains unfolded and inactive in aqueous solution (Figure 5). As a result, SVS-1 can selectively kill various cancer cell lines such as MCF-7, MDA-MB-436, KB, and A549 by lysing membranes, but showed low cytotoxicity to non-cancerous cells [39]. In addition, Chen et al. found that when PTP-7b (FLGALFKALSHLL), a therapeutic peptide, is attracted to the membrane and accumulated on cell surfaces, it self-assembles into exosome-like aggregates and induces membrane lysis, exhibiting a slightly different approach from other ACPs (Figure 6) [44].
Figure 4 Using scanning electron microscopy to observe the cell membrane in the drug-sensitive leukemia cell line K562 and its multi-drug resistant subline K562/ADM treated by polybia-MPI. The untreated K562 and K562/ADM showed a normal smooth surface (A, C). K562 and K562/ADM treated with 25 μM polybia-MPI showed a disrupted cell membrane (B, D) [38].
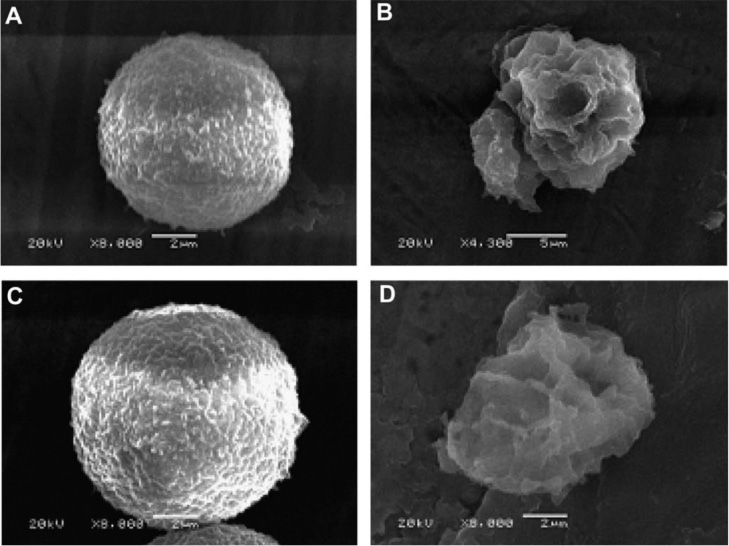
Figure 5 SVS-1 exists in solution as a random coil conformation. Once SVS-1 engages the cell membrane surface, it folds into a bioactive β-hairpin conformation, which can disrupt the cell membrane [39].
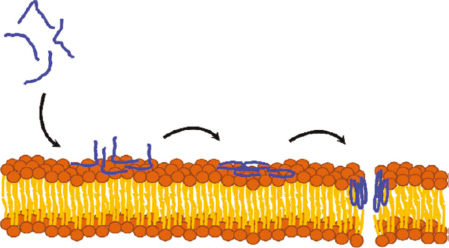
Figure 6 When the PTP-7b anticancer peptide reaches a certain concentration and comes into contact with the cell membrane, it will bind to the membrane and accumulate on cell surfaces, and then self-assemble into exosome-like aggregates and induce membrane lysis [44].
Effective drug transport into tumor cells remains a major problem in tumor therapy, due to the difficulty faced in passing the drug through the protective physiological barriers in tumor tissues. Although the selective permeability of cell membranes that allows the transport of bio-functional molecules is crucial in protecting cell integrity, it is also the greatest barrier that researchers face in drug delivery [45]. Consequently, it makes intracellular site-specific drug transport a very challenging task. Other than that, the strategy of drug penetration from blood vessels into tumors has yielded limited results as it may lead to uneven distribution of drugs to different parts of the tumor, rendering the drug ineffective or may even increase the likelihood of developing drug resistance in the tumor [46]. A group of peptides called the cell penetrating peptides, that have been demonstrated to be effective in delivering drugs into tumor cells, may address this problem [47]. As membrane active peptides, some researchers have proposed that ACPs and cell-penetrating peptides are highly similar in several aspects. For example, ACPs have a short amino sequence and contain cationic regions with a high affinity for cell membranes [15]. Therefore, similar to CCPs, they also improve the efficiency of drug delivery by promoting tumor penetration and improving drug distribution within tumors [48]. On the other hand, some ACPs act by membrane interaction mechanisms leading to membrane destabilization and enter the cell without destroying the integrity of the cell, which may be the major reason for its drug delivery function [49, 50].
ACPs improve drug delivery efficiency by co-administration with chemotherapy drugs or by modifying nanoparticles. It was noticed that some ACPs could penetrate the cell membrane at low concentrations but lysed the membrane at high concentrations. This piece of information indicates that the mechanism of the peptide may be tuned as intended by altering its concentration. NRC-03, NRC-07, gomesin, cecropin A, and DVD-1P are some of the examples of this type of ACP [51, 52]. Hilchie et al. established that the pleurocidin family of cationic antimicrobial peptides (NRC-03 and NRC-07) could kill multiple breast cancer cell lines by binding to negatively charged molecules on the cell membrane that subsequently lead to membrane lysis. In addition, both NRC-03 and NRC-07 could increase the chemosensitivity of breast cancer to cisplatin [53]. Gomesin is a potential anticancer peptide that was originally isolated from the hemocytes of a spider, Acanthoscurria gomesiana. It has been reported that gomesin could efficiently treat subcutaneous b16F10-NEX2 melanoma in mice by directly interacting with the cell membranes and at the same time, facilitate the delivery of drugs into tumor cells at low concentrations [54]. Moreover, tachyplesin, a cell penetrating antimicrobial peptide, was developed as a nanocarrier for anti-miR210A, which could efficiently deliver miRNA inside glioma cells cultured as two-dimensional (2D) and three-dimensional (3D) spheroid models [49]. In general, however, current research on ACPs for drug delivery is still limited.
Non-membrane perturbation mechanism of anticancer peptides
Even though membrane interaction was considered as the primary activity of ACPs, it has been proven in recent years that ACPs could also target the metabolism or cell division of cancer cells, in addition to affecting the immune system [55]. We hereby briefly summarize some mechanisms of ACPs that deviate from membrane perturbations, including angiogenesis inhibition, tumor apoptosis induction, and essential cell protein targeting or immune cell recruitment to attack cancer cells [37]. These effects are not common features of ACPs, but they play important synergistic roles in the anticancer effects of ACPs [56].
There are many ACPs such as P9, P12, and SP5031 that could efficiently inhibit angiogenesis and fight tumors by interfering with the interactions between growth factors and their receptors [57–59]. Some ACPs may bind to the cell surface and penetrate the cell membrane, while other non-lytic ACPs may simply pass through the membrane and enter the intracellular compartment, both of which can disrupt the mitochondria and cause programmed cell death [22]. Bovine lactoferricin (LfcinB), an ACP derived from cow’s milk, fights against cancer cells by inducing apoptosis in several cancer cells types, including MDA-MB-435 cells and THP-1 human monocytic leukemia cells [60–62]. It has been reported that some peptides could interfere with functional proteins involved in tumorigenesis and tumor progression to eradicate tumor cells. For example, the human anticancer peptide LL-37 can affect tumor development by inhibiting proteasome in gastric cancer cells [63, 64].
As mentioned already, another interesting anticancer mechanism is the subsequent effects caused by ACPs on the immune system after tumor cell lysis and the release of cellular contents. Such a mechanism is displayed by some ACPs such as LTX-315 and LTX-401 [65, 66]. Zhou et al. proved that the oncolytic peptide LTX-315 could cause transient focal necrosis accompanied by a large release of HMGB1 and ATP, as well as activation of caspase-3 in a fraction of the cells after being injected into the tumor. At the same time, LTX-315 could induce a local inflammatory response by the infiltration of T-lymphocytes and myeloid cells. These phenomena suggest the ability of LTX-315 in eliciting immunogenic cell death (ICD) [67]. Another oncolytic peptide LTX-401 can not only cause necrotic lysis of tumor cells, but also trigger some immunogenic events upon its injection into the tumor. It has been proven that LTX-401-mediated oncolysis could produce a synergistic effect when applied along with established immunotherapeutic regimens [66].
Problems faced by anticancer peptides as antitumor agents
As previously described, there have been many studies on the applications of ACPs, mainly due to their unique anticancer mechanism and the low tendency of developing drug resistance. Although many among the huge pool of naturally occurring and synthetic peptides exhibit potential anticancer effects, only a handful are being studied further for their clinical applications [68]. This is mainly due to the many obstacles faced by the development of ACPs as anticancer agents (Figure 7), such as high toxicity to normal mammalian cells [69]. Hence, researchers are working on ways to lower the toxicity level of ACPs to healthy mammalian cells while improving their tumor selectivity at the same time. Some studies have shown that arginine residues in cationic anticancer peptide interacted strongly with zwitterionic phospholipids, which may be the reason for ACPs’ high toxicity to normal cells [70, 71]. Due to this, some researchers anticipate that by using other cationic residues such as lysine to construct an ACP, which may still direct the binding of ACPs toward the negatively charged cells, they may avoid hemolysis [70]. There are also ACPs that are designed to only be activated at the tumor site but stayed inactive elsewhere in vivo, so as to reduce toxicity to normal cells. DVD-1P, a novel anticancer peptide designed by Shi and Schneider, contains a negatively charged phosphorylated tyrosine at the C-terminal region, which disrupted the amphiphilicity of the peptide and keeps the peptide unfolded and inactive. However, when it is dephosphorylated and subsequently binds to the negatively charged membranes, it will form an amphiphilic membrane-active conformation. Therefore, DVD-P1 selectively lysed cancer cells that expressed alkaline phosphatase (ALP) and had negatively charged cell membranes [51]. AMitP is a novel acid-activated ACPs that was constructed by combining MitP with its anionic peptide MitPE via a disulfide linker. AMitP showed remarkable anticancer activity in an acidic tumor microenvironment but low cytotoxicity in tissues with a normal pH [72]. Improving the tumor-targeting ability of ACPs by targeted modification is also a potential approach to lower cytotoxicity [73, 74]. Hu et al. designed a chimeric peptide HPRP-A1-iRGD by modifying cationic anticancer peptide HPRP-A1 with its tumor-homing/penetration domain (iRGD). This peptide displayed higher affinity and selectivity for tumors and stronger anti-tumor activity as compared to single HPRP-A1 [75].
Figure 7 Four major problems faced by the application of ACPs are shown here. (1) Cytotoxicity to normal cells, because normal cell membranes also have some negative charges. (2) Instability of ACPs in serum, resulting in low bioavailability. (3) Harmful anti-ACP immune responses including potentially dangerous allergic responses and the production of treatment-neutralizing antibodies. (4) High cost of peptide production.
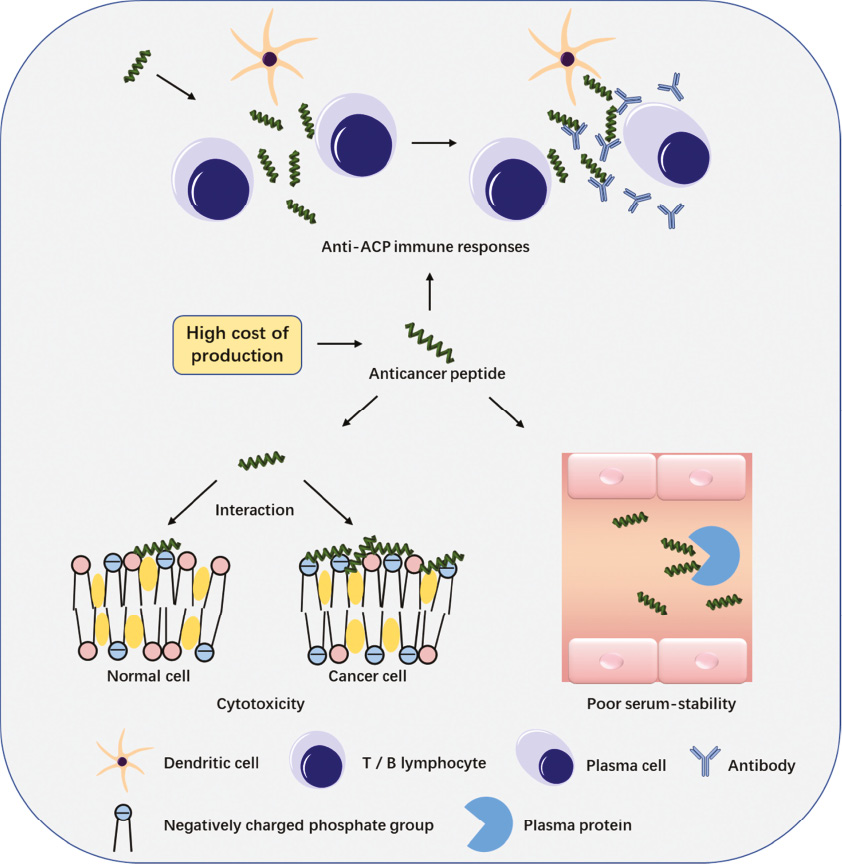
Another requirement for the clinical usage of ACPs in cancer treatment is their stability in serum. The presence of certain proteins including serum proteases and anionic serum proteins in serum may reduce the efficacy of ACPs [76]. For example, low-density lipoprotein (LDL) strongly inhibits cell lysis brought about by ACPs. Only a number of natural ACPs are very stable in serum; while synthetic ACPs are always designed as serum-stable peptides [35]. Thus, there are cases where ACPs are more stable against proteolytic degradation by serum protease after their naturally occurring amino acids are replaced with synthetic ones such as D-amino-acids [77–79]. The high cost of peptide production is also regarded as a major hurdle to the application of ACPs. However, protein- and peptide-based drugs such as hormone, antidiabetic and antihypertensive drugs have been used clinically for decades [80, 81]. Pharmaceutical companies invest heavily in the development of ACPs, which will inevitably lead to the technological progress and innovation strategy of the mass production of ACPs [22]. Other than that, it has to be borne in mind that ACPs are perceived as foreign substances in the human body, so they may incite harmful anti-ACP immune responses including potentially dangerous allergic responses and the production of treatment-neutralizing antibodies. To avoid this problem, either ACPs derived from humans can be opted for, or ACPs should be co-administered along with immunosuppressants [35]. Furthermore, ACPs could be delivered by nanosystems such as liposomes to the tumor site, in order to reduce direct contact between peptides and immune cells and thus avoiding the development of anti-ACP immunity [35, 82].
At present, great progress has been made in the research of anticancer peptides, with several ACPs (such as LTX-315, LL-37) already in clinical trials [19]. The human cathelicidin LL-37 is a cationic amphiphilic peptide that can directly eliminate bacteria by interacting with negatively charged cell membranes [63]. In addition to its antibacterial properties, LL-37 has some other functional attributes including anticancer, antifungal, antiviral, and immunomodulatory activities [83, 84]. However, the clinical trials for LL-37 are only limited to intratumoral administration [22]. Another ACP, LTX315 is a lactoferrin-derived lytic peptide that instigates tumor necrosis by lysing cancer cell and subsequently induces anti-tumor immunity. LTX-315 is currently undergoing phase I/II clinical trials for several types of solid tumors [85]. Although some anticancer peptides are already in clinical trials, the cytotoxicity to normal cells and instability in serum remain major obstacles for the clinical translation of ACPs [24]. Moreover, the phase I clinical trials of some ACPs suggest that the short half-life of the peptide was also a barrier that hinders the application of ACPs [24].
Despite posing great potential for practical application in anticancer treatments, solutions for the problems that have been described about ACPs should be identified before they can be widely used in reality [37]. Among the problems faced by ACPs, high toxicity to normal cells and low bioavailability due to serum instability are the major bottlenecks that are preventing ACPs from becoming cancer therapeutic agents. To overcome these problems, we can modify anticancer peptides in a combination of the following two aspects. On the one hand, we can make full use of the characteristics of tumor cells themselves and the tumor microenvironment, which are different from normal cells and tissues, and let the ACPs act specifically on tumor sites. For example, high expression of the ALP on the surface of some tumor cells and the slightly acidic environment of tumor sites [51, 86]. On the other hand, biomaterials can be used to modify anticancer peptides to increase their stability and prolong their half-life, such as nanoparticles. It is hoped that the bottleneck of the utilization of anticancer peptides can be overcome by using the advantages of biology and materials science simultaneously through biomedical engineering.
Future outlook
Conventional chemotherapy drugs pose many harmful side effects as they usually target all rapidly dividing cells, including normal cells in the body. In contrast, ACPs selectively interact with cancer cell membranes that are negatively charged, allowing a certain level of specificity. Besides, ACPs can also be used to treat tumors via some other mechanisms such as inducing tumor apoptosis, targeting essential cell proteins, or recruiting immune cells. Nonetheless, the exploration of ACPs’ potential in cancer treatment is still at a primitive stage and a considerable amount of research is needed to further clarify the exact mechanism by which ACPs interact with cancer cells. In addition, current chemotherapy drugs must enter cells to be effective, making it easier for cancer cells to develop resistance through several mechanisms such as efflux pumps. This confers an advantage to ACPs that act directly on cell membranes unlike conventional drugs. These advantages of ACPs have attracted the interest of researchers, leading to many ongoing studies that focus on the application of ACPs in cancer therapy, as outlined in this review. Nevertheless, ACPs pose many challenges that cannot be ignored if they are to be promoted as anticancer agents, such as high cytotoxicity, poor serum-stability, high cost of peptide productions, and harmful anti-ACP immune responses. Most ACPs remain in the research phase, with only a few in clinical trials. Scientists have proposed some corresponding solutions as already mentioned, but the usage of anticancer peptides in cancer treatment remains limited. One promising solution is nanosystems. Nanoparticles could effectively improve the selectivity of ACPs to tumors with the assistance of targeted modifications or tumor microenvironmental response strategies [87]. In addition, nanosystems such as liposomes and gold nanoparticles have been widely used in drug delivery systems due to their prolonged drug half-life, enhanced drug stability, and selective release of drugs [82]. Nanosystems can thus be used to deliver ACPs as an alternative strategy to overcome problems that are currently faced by ACPs. With more and more research focusing on overcoming these limitations and obstacles, ACPs look likely to bring about new opportunities for cancer prevention and treatment.
Conclusion
The development of drug resistance and harmful side effects are important reasons for the failure of any anticancer drug therapy. Therefore, there is an urgent need to develop new treatments that reduce the risk of drug resistance developing and increase cancer selectivity. ACPs, having been discovered for decades, fight against cancer by their unique mechanism of interacting with negatively charged cell membranes, so they are selective to cancer cells and do not easily develop drug resistance. However, only a few ACPs have been used in medical practice due to some of their own disadvantages, such as high toxicity to normal cells and low bioavailability. Currently, researchers have found a series of ways to overcome these problems, among which the construction of anticancer peptide nanosystems using biomedical engineering techniques is a promising approach. Anticancer peptide nanosystems can not only improve the tumor selectivity of anticancer peptides through targeted modification or tumor microenvironmental response strategy, but also improve its bioavailability. The advantages of biology and materials complement each other here, which will allow more and more anticancer peptides to be applied in the clinic.
Acknowledgments
This work was supported by grants from the Fundamental Research Funds for the Central Universities (20ykjc03), the National Science Foundation of China (81772837), Guangdong Natural Science Foundation (2018A0303130322, 2016A030313834 and 2017A030313871), the Science and Technology Foundation of the Guangdong Province (2019A050510016) and Guangdong Innovation and Entrepreneurship Team Projects (2019BT02Y198).
Conflict of interest
The authors declare no conflict of interest.
References
- Fidler MM. Bray F, Soerjomataram I. The global cancer burden and human development: a review. Scand J Public Health 2018;46: 27-36. [PMID: 28669281 DOI: 10.1177/1403494817715400]
- Hashim D, Boffetta P, La Vecchia C, Rota M, Beryuccio P, et al. The global decrease in cancer mortality: trends and disparities. Ann Oncol 2016;27:926-33. [PMID: 26802157 DOI: 10.1093/annonc/mdw027]
- Torre LA, Bray F, Siegal RL, Ferlay J, Lortet-Tieulent J, et al. Global Cancer Statistics, 2012. Cancer J Clin 2015;65:87-108. [PMID: 25651787 DOI: 10.3322/caac.21262]
- Gonen N, Assaraf YG. Antifolates in cancer therapy: structure, activity and mechanisms of drug resistance. Drug Resist Update 2012;15:183-210. [PMID: 22921318 DOI: 10.1016/j.drup.2012.07.002]
- Zhitomirsky B, Assaraf YG, Lysosomes as mediators of drug resistance in cancer. Drug Resist Update 2016;24:23-33. [PMID: 26830313 DOI: 10.1016/j.drup.2015.11.004]
- Martinez-Outschoorn UE, Peiris-Pagés M, Pestell RG, Sotgia F, Lisanti MP. Cancer metabolism: a therapeutic perspective. Nat Rev Clin Oncol 2017;14:11-31. [PMID: 27141887 DOI: 10.1038/nrclinonc.2016.60]
- Cao Y. Adipocyte and lipid metabolism in cancer drug resistance. J Clin Invest. 2019;129:3006-17. [PMID: 31264969 DOI: 10.1172/JCI127201]
- Amable L. Cisplatin resistance and opportunities for precision medicine. Pharmacol Res 2016;106:27-36. [PMID: 26804248 DOI: 10.1016/j.phrs.2016.01.001]
- Wijdeven RH, Pang B, Assaraf YG, Neefjes J. Old drugs, novel ways out: drug resistance toward cytotoxic chemotherapeutics. Drug Resist Updat 2016;28:65-81. [PMID: 27620955 DOI: 10.1016/j.drup.2016.07.001]
- Nikolaou M, Pavlopoulou A, Georgakilas AG, Kyrodimos E. The challenge of drug resistance in cancer treatment: a current overview. Clin Exp Metastasis 2018;35:309-18. [PMID: 29799080 DOI: 10.1007/s10585-018-9903-0]
- Gaspar D, Veiga AS, Castanho MA. From antimicrobial to anticancer peptides. A review. Front Microbiol 2013;4:294. [PMID: 24101917 DOI: 10.3389/fmicb.2013.00294]
- Deslouches B, Steckbeck JD, Craigo JK, Doi Y, Burns JL, et al. Engineered cationic antimicrobial peptides to overcome multidrug resistance by ESKAPE pathogens. Antimicrob Agents Chemother 2015;59:1329-33. [PMID: 25421473 DOI: 10.1128/AAC.03937-14]
- Qin Y, Qin ZD, Chen J, Cai CG, Li L, et al. From antimicrobial to anticancer peptides: the transformation of peptides. Recent Pat Anticancer Drug Discov 2019;14:70-84. [PMID: 30663573 DOI: 10.2174/1574892814666190119165157]
- Aaghaz S, Gohel V, Kamal A. Peptides as potential anticancer agents. Curr Top Med Chem 2019. 19:1491-11. [PMID: 30686254 DOI: 10.2174/1568026619666190125161517]
- Henriques ST, Melo MN, Castanho MA. Cell-penetrating peptides and antimicrobial peptides: how different are they? Biochem J 2006;399:1-7. [PMID: 16956326 DOI: 10.1042/BJ20061100]
- Baxter AA, Lay FT, Poon IKH, Kvansakul M, Hulett MD. Tumor cell membrane-targeting cationic antimicrobial peptides: novel insights into mechanisms of action and therapeutic prospects. Cell Mol Life Sci 2017;74:3809-25. [PMID: 28770291 DOI: 10.1007/s00018-017-2604-z]
- Henzler-Wildman KA, Martinez GV, Brown MF, Ramamoorthy A. Perturbation of the hydrophobic core of lipid bilayers by the human antimicrobial peptide LL-37. Biochemistry 2004;43: 8459-69. [PMID: 15222757 DOI: 10.1021/bi036284s]
- Xu H, Chen CX, Hu J, Zhou P, Zeng P, et al. Dual modes of antitumor action of an amphiphilic peptide A(9)K. Biomaterials 2013;34:2731-7. [PMID: 23352040 DOI: 10.1016/j.biomaterials.2012.12.039]
- Felicio MR, Silva ON, Gonçalves S, Santos NC, Franco OL. Peptides with dual antimicrobial and anticancer activities. Front Chem 2017;5:5. [PMID: 28271058 DOI: 10.3389/fchem.2017.00005]
- Pirtskhalava M, Gabrielian A, Cruz P, Griggs HL, Squires RB, et al. DBAASP v.2: an enhanced database of structure and antimicrobial/cytotoxic activity of natural and synthetic peptides. Nucleic Acids Res 2016;44:D1104-12. [PMID: 26578581 DOI: 10.1093/nar/gkv1174]
- Ciumac D, Gong H, Hu X, Lu JR. Membrane targeting cationic antimicrobial peptides. J Colloid Interface Sci 2019;537:163-85. [PMID: 30439615 DOI: 10.1016/j.jcis.2018.10.103]
- Deslouches B, Di YP. Antimicrobial peptides with selective antitumor mechanisms: prospect for anticancer applications. Oncotarget 2017;8:46635-51. [PMID: 28422728 DOI: 10.18632/oncotarget.16743]
- Soblosky L, Ramamoorthy A, Chen Z. Membrane interaction of antimicrobial peptides using E. coli lipid extract as model bacterial cell membranes and SFG spectroscopy. Chem Phys Lipids 2015;187:20-33. [PMID: 25707312 DOI: 10.1016/j.chemphyslip.2015.02.003]
- Li X, Yi L, Li Z, Lan X, Leung PH, et al. Mechanism of anticancer effects of antimicrobial peptides. J Fiber Bioeng Inform 2015;8: 25-36. [DOI: 10.3993/jfbi03201503]
- Yin LM, Edwards MA, Li J, Yip CM. Roles of hydrophobicity and charge distribution of cationic antimicrobial peptides in peptide-membrane interactions. J Biol Chem 2012;287:7738-45. [PMID: 22253439 DOI: 10.1074/jbc.M111.303602]
- Li Y, Xiang Q, Zhang Q, Huang Y, Su Z. Overview on the recent study of antimicrobial peptides: origins, functions, relative mechanisms and application. Peptides 2012;37:207-15. [PMID: 22800692 DOI: 10.1016/j.peptides.2012.07.001]
- Papo N, Shai Y. Host defense peptides as new weapons in cancer treatment. Cell Mol Life Sci 2005;62:784-90. [PMID: 15868403 DOI: 10.1007/s00018-005-4560-2]
- Järvå M, Lay FT, Phan TK, Humble C, Poon IKH, et al. X-ray structure of a carpet-like antimicrobial defensin-phospholipid membrane disruption complex. Nat Commun 2018;9:1962. [PMID: 29773800 DOI: 10.1038/s41467-018-04434-y]
- Yang L, Harroun TA, Weiss TM, Ding L, Huang HW. Barrel-stave model or toroidal model? A case study on melittin pores. Biophys J 2001;81:1475-85. [PMID: 11509361 DOI: 10.1016/S0006-3495(01)75802-X]
- Schweizer F. Cationic amphiphilic peptides with cancer-selective toxicity. Eur J Pharmacol 2009;625:190-4. [PMID: 19835863 DOI: 10.1016/j.ejphar.2009.08.043]
- Brogden KA. Antimicrobial peptides: pore formers or metabolic inhibitors in bacteria? Nat Rev Microbiol 2005;3:238-50. [PMID: 15703760 DOI: 10.1038/nrmicro1098]
- Henzler Wildman KA. Lee DK, Ramamoorthy A. Mechanism of lipid bilayer disruption by the human antimicrobial peptide, LL-37. Biochemistry 2003;42:6545-58. [PMID: 12767238 DOI: 10.1021/bi0273563]
- Kenis H, Reutelingsperger C. Targeting phosphatidylserine in anti-cancer therapy. Curr Pharm Des 2009;15:2719-23. [PMID: 19689342 DOI: 10.2174/138161209788923903]
- Leite ML, da Cunha NB, Costa FF. Antimicrobial peptides, nanotechnology, and natural metabolites as novel approaches for cancer treatment. Pharmacol Ther 2018;183:160-76. [PMID: 29024740 DOI: 10.1016/j.pharmthera.2017.10.010]
- Hoskin DW, Ramamoorthy A. Studies on anticancer activities of antimicrobial peptides. Biochim Biophys Acta 2008;1778:357-75. [PMID: 18078805 DOI: 10.1016/j.bbamem.2007.11.008]
- Leite NB, Aufderhorst-Roberts A, Palma MS, Connell SD, Ruggiero Neto J, et al. PE and PS lipids synergistically enhance membrane poration by a peptide with anticancer properties. Biophys J 2015;109:936-47. [PMID: 26331251 DOI: 10.1016/j.bpj.2015.07.033]
- Wu D, Gao Y, Qi Y, Chen L, Ma Y, et al. Peptide-based cancer therapy: opportunity and challenge. Cancer Lett 2014;351:13-22. [PMID: 24836189 DOI: 10.1016/j.canlet.2014.05.002]
- Wang KR, Yan JX, Zhang BZ, Song JJ, Jia PF, et al. Novel mode of action of polybia-MPI, a novel antimicrobial peptide, in multi-drug resistant leukemic cells. Cancer Lett 2009;278:65-72. [PMID: 19233550 DOI: 10.1016/j.canlet.2008.12.027]
- Sinthuvanich C, Veiga AS, Gupta K, Gaspar D, Blumenthal R, et al. Anticancer beta-hairpin peptides: membrane-induced folding triggers activity. J Am Chem Soc 2012;134:6210-7. [PMID: 22413859 DOI: 10.1021/ja210569f]
- Roudi R, Syn NL, Roudbary M. Antimicrobial peptides as biologic and immunotherapeutic agents against cancer: a comprehensive overview. Front Immunol 2017;8:1320. [PMID: 29081781 DOI: 10.3389/fimmu.2017.01320]
- Soliman C, Eastwood S, Truong VK, Ramsland PA, Elbourne A. The membrane effects of melittin on gastric and colorectal cancer. PLoS One 2019;14:e0224028. [PMID: 31622415 DOI: 10.1371/journal.pone.0224028]
- Lehmann J, Retz M, Sidhu SS, Suttmann H, Sell M, et al. Antitumor activity of the antimicrobial peptide magainin II against bladder cancer cell lines. Eur Urol 2006;50:141-7. [PMID: 16476519 DOI: 10.1016/j.eururo.2005.12.043]
- Wang KR, Zhang BZ, Zhang W, Yan JX, Li J, et al. Antitumor effects, cell selectivity and structure-activity relationship of a novel antimicrobial peptide polybia-MPI. Peptides 2008;29:963-8. [PMID: 18328599 DOI: 10.1016/j.peptides.2008.01.015]
- Chen L, Patrone N, Liang JF. Peptide self-assembly on cell membranes to induce cell lysis. Biomacromolecules 2012;13:3327-33. [PMID: 22934601 DOI: 10.1021/bm301106p]
- Raucher D, Ryu JS. Cell-penetrating peptides: strategies for anticancer treatment. Trends Mol Med 2015;21:560-70. [PMID: 26186888 DOI: 10.1016/j.molmed.2015.06.005]
- Primeau AJ, Rendon A, Hedley D, Lilge L, Tannock IF. The distribution of the anticancer drug Doxorubicin in relation to blood vessels in solid tumors. Clin Cancer Res 2005;11:8782-8. [PMID: 16361566 DOI: 10.1158/1078-0432.CCR-05-1664]
- Guidotti G, Brambilla L, Rossi D. Cell-penetrating peptides: from basic research to clinics. Trends Pharmacol Sci 2017;38:406-24. [PMID: 28209404 DOI: 10.1016/j.tips.2017.01.003]
- Sugahara KN, Teesalu T, Karmali PP, Kotamraju VR, Agemy L, et al., Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science 2010;328:1031-5. [PMID: 20378772 DOI: 10.1126/science.1183057]
- Jana A, Narula P, Chugh A, Kulshreshtha R. Efficient delivery of anti-miR-210 using Tachyplesin, a cell penetrating peptide, for glioblastoma treatment. Int J Pharm 2019;572:118789. [PMID: 31726199 DOI: 10.1016/j.ijpharm.2019.118789]
- Zhang L, Rozek A, Hancock RE. Interaction of cationic antimicrobial peptides with model membranes. J Biol Chem 2001:276: 35714-22. [PMID: 11473117 DOI: 10.1074/jbc.M104925200]
- Shi J, Schneider JP. De novo design of selective membrane-active peptides by enzymatic control of their conformational bias on the cell surface. Angew Chem Int Ed Engl 2019;58:13706-10. [PMID: 31268617 DOI: 10.1002/anie.201902470]
- Hui L, Leung K, Chen HM. The combined effects of antibacterial peptide cecropin A and anti-cancer agents on leukemia cells. Anticancer Res 2002;22:2811-6. [PMID: 12530001]
- Hilchie AL, Doucette CD, Pinto DM, Patrzykat A, Douglas S, et al. Pleurocidin-family cationic antimicrobial peptides are cytolytic for breast carcinoma cells and prevent growth of tumor xenografts. Breast Cancer Res 2011;13:R102. [PMID: 22023734 DOI: 10.1186/bcr3043]
- Rodrigues EG, Dobroff AS, Cavarsan CF, Paschoalin T, Nimrichter L, et al. Effective topical treatment of subcutaneous murine B16F10-Nex2 melanoma by the antimicrobial peptide gomesin. Neoplasia 2008;10:61-8. [PMID: 18231639 DOI: 10.1593/neo.07885]
- Hancock RE, Haney EF, Gill EE. The immunology of host defence peptides: beyond antimicrobial activity. Nat Rev Immunol 2016;16:321-34. [PMID: 27087664 DOI: 10.1038/nri.2016.29]
- Do N, Weindl G, Grohmann L, Salwiczek M, Koksch B, et al. Cationic membrane-active peptides–anticancer and antifungal activity as well as penetration into human skin. Exp Dermatol 2014;23: 326-31. [PMID: 24661024 DOI: 10.1111/exd.12384]
- Wu X, Huang H, Wang C, Lin S, Huang Y, et al. Identification of a novel peptide that blocks basic fibroblast growth factor-mediated cell proliferation. Oncotarget 2013;4:1819-28. [PMID: 24142482 DOI: 10.18632/oncotarget.1312]
- Wang W, Chen X, Li T, Li Y, Wang R, et al. Screening a phage display library for a novel FGF8b-binding peptide with anti-tumor effect on prostate cancer. Exp Cell Res 2013;319:1156-64. [PMID: 23466786 DOI: 10.1016/j.yexcr.2013.02.007]
- Lee E, Koskimaki JE, Pandey NB, Popel AS, et al. Inhibition of lymphangiogenesis and angiogenesis in breast tumor xenografts and lymph nodes by a peptide derived from transmembrane protein 45A. Neoplasia 2013;15:112-24. [PMID: 23441126 DOI: 10.1593/neo.121638]
- Zhang TN, Liu N. Effect of bovine lactoferricin on DNA methyltransferase 1 levels in Jurkat T-leukemia cells. J Dairy Sci 2010;93:3925-30. [PMID: 20723665 DOI: 10.3168/jds.2009-3024]
- Furlong SJ, Mader JS, Hoskin DW. Lactoferricin-induced apoptosis in estrogen-nonresponsive MDA-MB-435 breast cancer cells is enhanced by C6 ceramide or tamoxifen. Oncol Rep 2006;15: 1385-90. [PMID: 16596215]
- Furlong SJ, Ridgway ND, Hoskin DW. Modulation of ceramide metabolism in T-leukemia cell lines potentiates apoptosis induced by the cationic antimicrobial peptide bovine lactoferricin. Int J Oncol 2008;32:537-44. [PMID: 18292930]
- Kuroda K, Okumura K, Isogai H, Isogai E. The human cathelicidin antimicrobial peptide LL-37 and mimics are potential anticancer drugs. Front Oncol 2015;5:144. [PMID: 26175965 DOI: 10.3389/fonc.2015.00144]
- Wu WKK, Sung JJY, To KF, Yu L, Li HT, et al. The host defense peptide LL-37 activates the tumor-suppressing bone morphogenetic protein signaling via inhibition of proteasome in gastric cancer cells. J Cell Physiol 2010;223:178-86. [PMID: 20054823 DOI: 10.1002/jcp.22026]
- Sveinbjornsson B, Camilio KA, Haug BE, Rekdal Ø. LTX-315: a first-in-class oncolytic peptide that reprograms the tumor microenvironment. Future Med Chem 2017;9:1339-44. [PMID: 28490192 DOI: 10.4155/fmc-2017-0088]
- Xie W, Mondragón L, Mauseth B, Wang Y, Pol J, et al. Tumor lysis with LTX-401 creates anticancer immunity. Oncoimmunology 2019;8:1594555. [PMID: 31143516 DOI: 10.1080/2162402X.2019.1594555]
- Zhou H, Forveille S, Sauvat A, Yamazaki T, Senovilla L, et al. The oncolytic peptide LTX-315 triggers immunogenic cell death. Cell Death Dis 2016;7:e2134. [PMID: 26962684 DOI: 10.1038/cddis.2016.47]
- Gordon YJ, Romanowski EG, McDermott AM. A review of antimicrobial peptides and their therapeutic potential as anti-infective drugs. Curr Eye Res 2005;30:505-15. [PMID: 16020284 DOI: 10.1080/02713680590968637]
- Xiao YF, Jie MM, Li BS, Hu CJ, Xie R, et al. Peptide-based treatment: a promising cancer therapy. J Immunol Res 2015;2015:761820. [PMID: 26568964 DOI: 10.1155/2015/761820]
- Giuliani A, Pirri G, Nicoletto S. Antimicrobial peptides: an overview of a promising class of therapeutics. Cent Eur J Biol 2007;2: 1-33. [DOI: 10.2478/s11535-007-0010-5]
- Yang ST, Shin SY, Lee CW, Kim Y-C, Hahm K-S, et al. Selective cytotoxicity following Arg-to-Lys substitution in tritrpticin adopting a unique amphipathic turn structure. FEBS Lett 2003;540:229-33. [PMID: 12681513 DOI: 10.1016/s0014-5793(03)00266-7]
- Song J, Zhang W, Kai M, Chen J, Liang R, et al. Design of an acid-activated antimicrobial peptide for tumor therapy. Mol Pharm 2013;10:2934-41. [PMID: 23819484 DOI: 10.1021/ mp400052s]
- Huang Y, Feng Q, Yan Q, Hao X, Chen Y, et al. Alpha-helical cationic anticancer peptides: a promising candidate for novel anticancer drugs. Mini Rev Med Chem 2015;15:73-81. [PMID: 25382016 DOI: 10.2174/1389557514666141107120954]
- Nemudraya AA, Makartsova AA, Fomin AS, Nushtaeva AA, Koval OA, et al. Tumor-specific peptide, selected from a phage peptide library, enhances antitumor activity of lactaptin. PLoS One 2016;11:e0160980. [PMID: 27513518 DOI: 10.1371/journal.pone.0160980]
- Hu C, Huang Y, Chen Y. Targeted modification of the cationic anticancer peptide HPRP-A1 with iRGD to improve specificity, penetration, and tumor-tissue accumulation. Mol Pharm 2019;16:561-72. [PMID: 30592418 DOI: 10.1021/acs.molpharmaceut.8b00854]
- Nguyen LT, Chau JK, Perry NA, de Boer L, Zaat SAJ, et al. Serum stabilities of short tryptophan- and arginine-rich antimicrobial peptide analogs. PLoS One 2010;5:e12684. [PMID: 20844765 DOI: 10.1371/journal.pone.0012684]
- Uggerhoj LE, Poulsen TJ, Munk JK, Fredborg M, Esben T, et al. Rational design of alpha-helical antimicrobial peptides: do’s and don’ts. Chembiochem 2015;16:242-53. [PMID: 25530580 DOI: 10.1002/cbic.201402581]
- de la Fuente-Nunez C, Reffuveille F, Mansour SC, Reckseidler- Zenteno SL, Hernández D, et al. D-enantiomeric peptides that eradicate wild-type and multidrug-resistant biofilms and protect against lethal Pseudomonas aeruginosa infections. Chem Biol 2015;22:196-205. [PMID: 25699603 DOI: 10.1016/j.chembiol.2015.01.002]
- Riedl S, Zweytick D, Lohner K. Membrane-active host defense peptides – challenges and perspectives for the development of novel anticancer drugs. Chem Phys Lipids 2011;164:766-81. [PMID: 21945565 DOI: 10.1016/j.chemphyslip.2011.09.004]
- Fosgerau K, Hoffmann T. Peptide therapeutics: current status and future directions. Drug Discov Today 2015;20:122-8. [PMID: 25450771 DOI: 10.1016/j.drudis.2014.10.003]
- Farrar D, Tuffnell DJ, West J, West HM. Continuous subcutaneous insulin infusion versus multiple daily injections of insulin for pregnant women with diabetes. Cochrane Database Syst Rev 2016;6:Cd005542. [PMID: 29545258 DOI: 10.1530/EJE-17-0804]
- Zhang C, Yang M, Ericsson AC. Antimicrobial peptides: potential application in liver cancer. Front Microbiol 2019;10:1257. [PMID: 31231341 DOI: 10.3389/fmicb.2019.01257]
- Chuang CM, Monie A, Wu A, Mao CP, Hung CF. Treatment with LL-37 peptide enhances antitumor effects induced by CpG oligodeoxynucleotides against ovarian cancer. Hum Gene Ther 2009;20:303-13. [PMID: 19272013 DOI: 10.1089/hum.2008.124]
- Tomasinsig L, De Conti G, Skerlavaj B, Piccinini R, Mazzilli M, et al. Broad-spectrum activity against bacterial mastitis pathogens and activation of mammary epithelial cells support a protective role of neutrophil cathelicidins in bovine mastitis. Infect Immun 2010;78:1781-8. [PMID: 20100862 DOI: 10.1128/IAI.01090-09]
- A Phase I, Open-label, multi-arm, multicentre, multi-dose, dose escalation study of LTX-315 as monotherapy or in combination with either ipilimumab or pembrolizumab in patients with transdermally accessible tumours. 2019. https://adisinsight.springer.com/trials/700238936#disabled
- Liu J, Huang Y, Kumar A, Tan A, Jin S, et al. pH-sensitive nano-systems for drug delivery in cancer therapy. Biotechnol Adv 2014;32:693-710. [PMID: 24309541 DOI: 10.1016/j.biotechadv.2013.11.009]
- Shi J, Kantoff PW, Wooster R, Farokhzad OC. Cancer nanomedicine: progress, challenges and opportunities. Nat Rev Cancer 2017;17:20-37. [PMID: 27834398 DOI: 10.1038/nrc.2016.108]
- Vandamme D, Landuyt B, Luyten W, Schoofs L. A comprehensive summary of LL-37, the factotum human cathelicidin peptide. Cell Immunol 2012;280:22-35. [PMID: 23246832 DOI: 10.1016/j.cellimm.2012.11.009]
- Rozek A, Powers J-P, Friedrich CL, Hancock RE. Structure-based design of an indolicidin peptide analogue with increased protease stability. Biochemistry 2003;42:14130-8. [PMID: 14640680 DOI: 10.1021/bi035643g]
- Hristova K, Dempsey CE, White SH. Structure, location, and lipid perturbations of melittin at the membrane interface. Biophys J 2001;80:801-11. [PMID: 11159447 DOI: 10.1016/S0006-3495(01)76059-6]
- Sharma SV. Melittin-induced hyperactivation of phospholipase A2 activity and calcium influx in ras-transformed cells. Oncogene 1993;8:939-47. [PMID: 8455945]
- Saini SS, Chopra AK, Peterson JW. Melittin activates endogenous phospholipase D during cytolysis of human monocytic leukemia cells. Toxicon 1999;37:1605-19. [PMID: 10708799 DOI: 10.1016/s0041-0101(99)00219-6]
- Killion JJ, Dunn JD. Differential cytolysis of murine spleen, bone-marrow and leukemia cells by melittin reveals differences in membrane topography. Biochem Biophys Res Commun 1986;139:222-7. [PMID: 3767954 DOI: 10.1016/s0006-291x(86)80102-4]
- Midoux P, Mayer R, Monsigny M. Membrane permeabilization by alpha-helical peptides: a flow cytometry study. Biochim Biophys Acta 1995;1239:249-56. [PMID: 7488630 DOI: 10.1016/0005-2736(95)00163-W]
- Wang K, Yan J, Liu X, Zhang J, Chen R, et al. Novel cytotoxity exhibition mode of polybia-CP, a novel antimicrobial peptide from the venom of the social wasp Polybia paulista. Toxicology 2011;288:27-33. [PMID: 21745529 DOI: 10.1016/j.tox.2011.06.014]
- Iwasaki T, Ishibashi J, Tanaka H, Sato M, Asaoka A, et al. Selective cancer cell cytotoxicity of enantiomeric 9-mer peptides derived from beetle defensins depends on negatively charged phosphatidylserine on the cell surface. Peptides 2009;30:660-8. [PMID: 19154767 DOI: 10.1016/j.peptides.2008.12.019]
- Cruz-Chamorro L, Puertollano MA, Puertollano E, de Cienfuegos GA, de Pablo MA. In vitro biological activities of magainin alone or in combination with nisin. Peptides 2006;27:1201-9. [PMID: 16356589 DOI: 10.1016/j.peptides.2005.11.008]
- Takeshima K, Chikushi A, Lee K-K, YoneharaS, Matsuzaki K. Translocation of analogues of the antimicrobial peptides magainin and buforin across human cell membranes. J Biol Chem 2003;278:1310-5. [PMID: 12417587 DOI: 10.1074/jbc.M208762200]
- Gaspar D, Veiga AS, Sinthuvanich C, Schneider JP, Castanho MARB. Anticancer peptide SVS-1: efficacy precedes membrane neutralization. Biochemistry 2012;51:6263-5. [PMID: 22839778 DOI: 10.1021/bi300836r]
- Yoo YC, Watanabe R, Koikea Y, Mitobe M, Shimazaki KI, et al. Apoptosis in human leukemic cells induced by lactoferricin, a bovine milk protein-derived peptide: involvement of reactive oxygen species. Biochem Biophys Res Commun 1997;237:624-8. [PMID: 9299415 DOI: 10.1006/bbrc.1997.7199]
- Eliassen LT, Berge G, Sveinbjørnsson B, Svendsen JS, Vorland LH, et al. Evidence for a direct antitumor mechanism of action of bovine lactoferricin. Anticancer Res 2002;22:2703-10. [PMID: 12529985]
- Mader JS, Salsman J, Conrad DM, Hoskin DW. Bovine lactoferricin selectively induces apoptosis in human leukemia and carcinoma cell lines. Mol Cancer Ther 2005;4:612-24. [PMID: 15827335 DOI: 10.1158/1535-7163.MCT-04-0077]
- Eliassen LT, Berge G, Leknessund A, Wikman M, Lindin I, et al. The antimicrobial peptide, lactoferricin B, is cytotoxic to neuroblastoma cells in vitro and inhibits xenograft growth in vivo. Int J Cancer 2006;119:493-500. [PMID: 16572423 DOI: 10.1002/ijc.21886]
- Pan WR, Chen PW, Chen YLS, Hsu HC, Lin CC, et al. Bovine lactoferricin B induces apoptosis of human gastric cancer cell line AGS by inhibition of autophagy at a late stage. J Dairy Sci 2013;96:7511-20. [PMID: 24140317 DOI: 10.3168/jds.2013-7285]
- Okumura K, Itoh A, Isogai E, Hirose K, Hosokawa Y, et al. C-terminal domain of human CAP18 antimicrobial peptide induces apoptosis in oral squamous cell carcinoma SAS-H1 cells. Cancer Lett 2004;212:185-94. [PMID: 15279899 DOI: 10.1016/j.canlet.2004.04.006]
- Mader JS, Mookherjee N, Hancock REW, Bleackley RC. The human host defense peptide LL-37 induces apoptosis in a calpain- and apoptosis-inducing factor-dependent manner involving Bax activity. Mol Cancer Res 2009;7:689-702. [PMID: 19435812 DOI: 10.1158/1541-7786.MCR-08-0274]
- Haug BE, Camilio KA, Eliassen LT, Stensen W, Svendsen JS, et al. Discovery of a 9-mer cationic peptide (LTX-315) as a potential first in class oncolytic peptide. J Med Chem 2016;59:2918-27. [PMID: 26982623 DOI: 10.1021/acs.jmedchem.5b02025]
- Camilio KA, Wang MY, Mauseth B, Waagene S, Kvalheim G, et al. Combining the oncolytic peptide LTX-315 with doxorubicin demonstrates therapeutic potential in a triple-negative breast cancer model. Breast Cancer Res 2019;21:9. [PMID: 30670061 DOI: 10.1186/s13058-018-1092-x]
- Forveille S, Zhou H, Sauvat A, Bezu L, Müller K, et al. The oncolytic peptide LTX-315 triggers necrotic cell death. Cell Cycle 2015;14:3506-12. [PMID: 26566869 DOI: 10.1080/15384101.2015.1093710]
- Camilio KA, Berge G, Ravuri CS, Rekdal O, Sveinbjørnsson B. Complete regression and systemic protective immune responses obtained in B16 melanomas after treatment with LTX-315. Cancer Immunol Immunother 2014;63: 601-13. [PMID: 24676901 DOI: 10.1007/s00262-014- 1540-0]
- Nestvold J, Wang MY, Camilio KA, Zinöcker S, Elisabeth Tjelle T, et al. Oncolytic peptide LTX-315 induces an immune-mediated abscopal effect in a rat sarcoma model. Oncoimmunology 2017;6:e1338236. [PMID: 28920000 DOI: 10.1080/2162402X.2017.1338236]
- Han Y, Cui Z, Li YH, Hsu W-H, Lee BH. In vitro and in vivo anticancer activity of pardaxin against proliferation and growth of oral squamous cell carcinoma. Mar Drugs 2015;14:2. [PMID: 26703631 DOI: 10.3390/md14010002]