Albinism-Associated Genes and Non-Skin Cancer: Bioinformatics Analysis of Relevant miRNAs
1Department of Nursing, University of Thessaly, Gaiopolis, Larissa 41500, Greece
2Department of Physiology, Faculty of Medicine, University of Thessaly, Biopolis, Larissa 41500, Greece
*Correspondence to: Erasmia Rouka, Department of Nursing, University of Thessaly, Gaiopolis, Larissa 41500, Greece, E-mail: errouka@uth.gr
Received: September 8 2025; Revised: December 2 2025; Accepted: December 24 2025; Published Online: February 13 2026
Cite this paper:
Cekani A, Gkertsou Z, Gkogko M et al. Albinism-Associated Genes and Non-Skin Cancer: Bioinformatics Analysis of Relevant miRNAs. BIO Integration 2026; 7: 1–2.
DOI: 10.15212/bioi-2025-0163. Available at: https://bio-integration.org/
Download citation
© 2026 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
The relevance of albinism to non-skin malignancies has not been sufficiently explored. Therefore, to extend analysis beyond the expected phenotypes, we focused on ten genes (MITF, MC1R, TYRP1, TYR, OCA2, DCT, LRMDA, SLC24A5, SLC45A2, and GPR143) associated with albinism, for which relevant information was obtained from the Online Catalog of Human Genes and Genetic Disorders (OMIM) database (https://omim.org/) [1].
Our in silico approach comprised four steps. The first step, interaction network prediction for each of the investigated genes, was conducted with the following filters: Homo sapiens, as many as 50 interactors, and medium confidence (score ≥ 0.400) in the STRING V.12 resource [2]. Subsequently, we entered each interactome into the FunRich software tool [3] for miRNA prediction, then conducted functional enrichment analysis relative to biological processes (BPs), sites of expression, and transcription factors (TFs) of post-transcriptional regulators targeting at least two of the investigated albinism genes. The third step involved disease enrichment of the predicted miRNAs with default settings in the RNADisease V4.0 repository [4]. Finally, to validate the associations of enriched miRNA-cancer types, we surveyed strong experimental evidence via the repository’s batch search option [4]. The selection of bioinformatic tools was based primarily on the quality of their documentation. Through STRING analysis, the 50 interactors with the highest confidence for each of the ten investigated albinism-associated genes were retrieved. The ten predicted networks shared 60 interactors. FunRich analysis identified several miRNAs involved in post-transcriptional regulation: 111 for OCA2, 113 for DCT, 87 for GPR1-43, 70 for LRMDA, 126 for MC1R, 194 for MITF, 147 for SLC24A5, 98 for TYR, 135 for TYRP1, and 147 for SLC45A2. The top five enriched Gene Ontology annotations for the identified miRNAs were the BPs regulation of translation, transport, cell communication, signal transduction, and nucleic acid metabolism; the expression sites skeletal muscle, heart, kidney, placenta, and brain; and the TFs MEF2A, POU2F1, SP4, SP1, and EGR1. The enriched BP annotations have been reported to play important roles in carcinogenesis. Among expression sites, the skin and eyes were expected to be present yet were not among the top five enriched annotations. This notable finding might indicate a complex regulatory shift between miRNAs and their mRNA targets, such that post-transcriptional regulators are expressed in the identified specialized tissues and organs, whereas their targets are abundant in other cell types. These findings might potentially guide future research toward tissue specificity. Moreover, several of the identified TFs have been shown to contribute to carcinogenesis.
Breast cancer, hepatocellular carcinoma, esophageal cancer, gastric cancer, pancreatic cancer, and glioblastoma were among the most enriched diseases (Figure 1). The search for strong experimental data verified the association between the enriched cancer types and the list of predicted post-transcriptional regulators. The evidence was particularly strong for breast cancer, hepatocellular carcinoma, and pancreatic cancer. Albinism, skin cancer, and non-skin malignancies could cluster via shared genes and pathways regulating cell division, growth, metabolism, and apoptosis.
Figure 1 Top 20 enriched annotations for diseases associated with miRNAs regulating the expression of the investigated albinism genes, as retrieved from RNADisease.
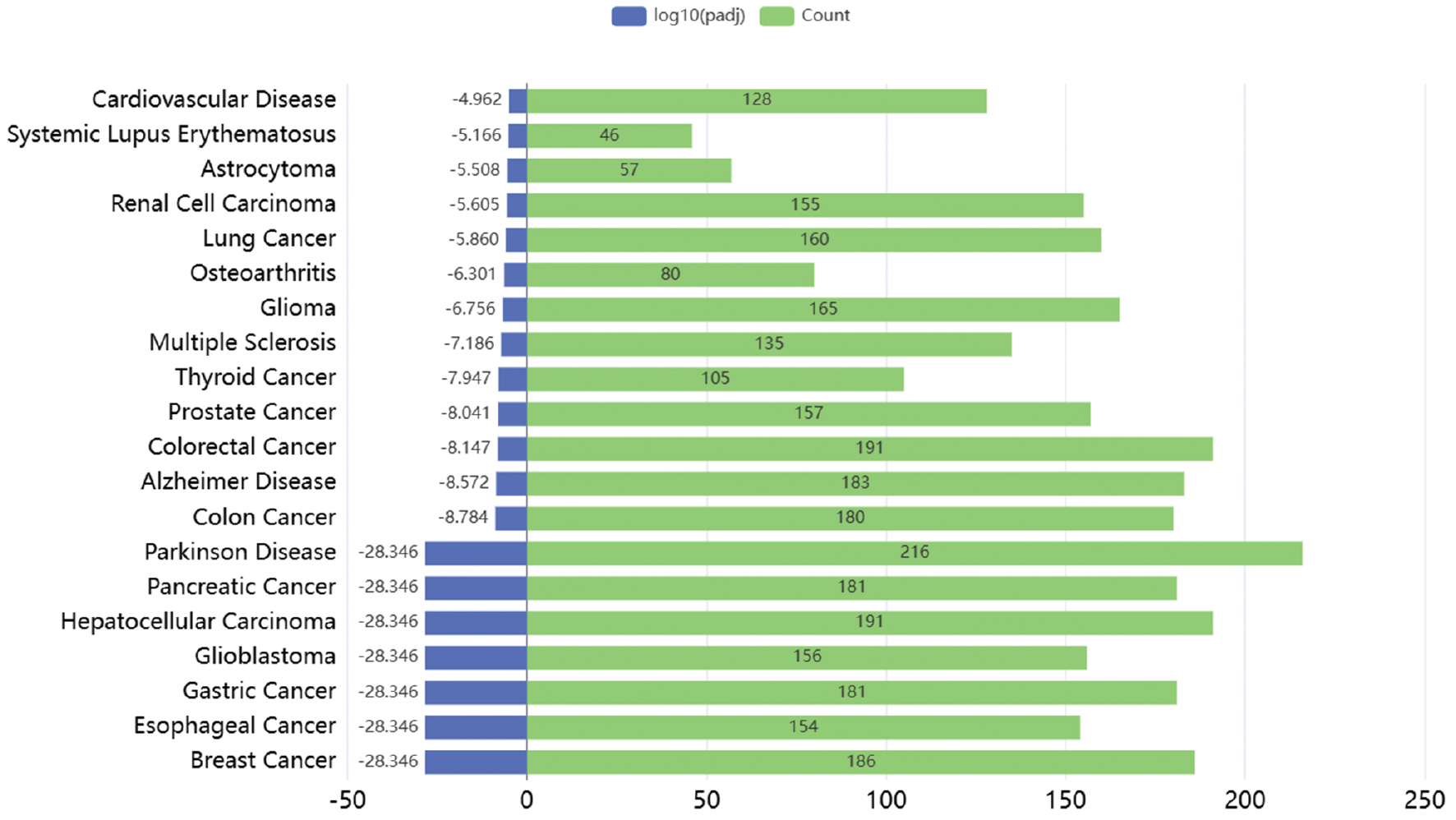
Collectively, although our findings relied solely on bioinformatic predictions, they lay a groundwork for evidence-based experimental research on specific candidate albinism-associated gene regulatory networks in non-skin cancer. Future investigations should verify miRNA-target gene interactions and determine whether these miRNAs play oncogenic or tumor-suppressive roles in each identified non-skin cancer type.
Data availability statement
The authors confirm that the data supporting the findings of this study are available from the corresponding author upon reasonable request.
Ethics statement
No direct interactions with human or animal subjects were involved. Therefore, ethical approval and informed consent were not required.
Author contributions
Anita Cekani: Investigation, Data curation, Writing—Original draft preparation. Zoi Gkertsou: Investigation, Data curation, Writing—Original draft preparation. Maria Gkogko: Investigation, Data curation, Writing—Original draft preparation. Dimitra Koutsogianni: Investigation, Data curation, Writing—Original draft preparation. Polyxeni Polychroniou: Investigation, Data curation, Writing—Original draft preparation. Christina-Markella Zidrou: Investigation, Data curation, Writing—Original draft preparation. Sotirios Zarogiannis: Writing—Review & Editing. Erasmia Rouka: Conceptualization, Validation, Writing—Review & Editing, Supervision.
Funding
This research received no external funding.
Conflict of interest
The authors declare that there are no conflicts of interest.
References
- Hamosh A, Amberger JS, Bocchini C, Scott AF, Rasmussen SA. Online Mendelian Inheritance in Man (OMIM®): Victor McKusick’s magnum opus. Am J Med Genet A 2021;185(11):3259-65. [PMID: 34169650 DOI: 10.1002/ajmg.a.62407]
- Szklarczyk D, Nastou K, Koutrouli M, Kirsch R, Mehryary F, et al. The STRING database in 2025: protein networks with directionality of regulation. Nucleic Acids Res 2025;53(D1):D730-7. [PMID: 39558183 DOI: 10.1093/nar/gkae1113]
- Fonseka P, Pathan M, Chitti SV, Kang T, Mathivanan S. FunRich enables enrichment analysis of OMICs datasets. J Mol Biol 2021;433(11):166747. [PMID: 33310018 DOI: 10.1016/j.jmb.2020.166747]
- Chen J, Lin J, Hu Y, Ye M, Yao L, et al. RNADisease v4.0: an updated resource of RNA-associated diseases, providing RNA disease analysis, enrichment and prediction. Nucleic Acids Res 2023;51(D1):D1397-404. [PMID: 36134718 DOI: 10.1093/nar/gkac814]