Effects of the Intestinal Microbiome and Metabolites on Neoadjuvant Chemotherapy Efficacy in Breast Cancer
1Department of Breast Surgery, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, PR China
2The First Clinical Medicine College, Nanjing Medical University, Nanjing, Jiangsu, PR China
3Department of Oncology, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, PR China
4Department of Physiology, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, PR China
5Department of General Surgery, The Affiliated Taizhou People’s Hospital of Nanjing Medical University, Taizhou School of Clinical Medicine, Nanjing Medical University, Taizhou, Jiangsu, PR China
aJingyue Fu and Hongxin Lin contributed equally to this work.
*Corresponding to: Yichao Zhu, School of Basic Medical Sciences, Nanjing Medical University, Nanjing, Jiangsu, PR China, E-mail: zhuyichao@njmu.edu.cn; Tiansong Xia, The First Affiliated Hospital of Nanjing Medical University, Nanjing, Jiangsu, PR China, E-mail: xiatsswms@163.com
Received: February 21 2025; Revised: April 8 2025; Accepted: June 6 2025; Published Online: July 9 2025
Cite this paper:
Fu J, Lin H, Li S et al. Effects of the Intestinal Microbiome and Metabolites on Neoadjuvant Chemotherapy Efficacy in Breast Cancer. BIO Integration 2025; 6: 1–14.
DOI: 10.15212/bioi-2025-0040. Available at: https://bio-integration.org/
Download citation
© 2025 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Background: Imbalances in the intestinal microbiome are closely associated with the occurrence and development of cancer, and can affect tumorigenesis by influencing the inflammatory response, regulating the immune system, producing specific metabolites, and participating in tumor signaling pathways.
Methods: This study investigated the relationships among intestinal microbial dynamics, metabolite profiles, and neoadjuvant chemotherapy (NAC) outcomes in patients with breast cancer. Patients were stratified by Miller-Payne (MP) grade into good (MP 4–5) or poor (MP 1–3) responders. Fecal samples from patients (pre- and post-NAC) were analyzed via 16S rRNA sequencing and untargeted metabolic analysis.
Results: After neoadjuvant chemotherapy, the species diversity and abundance of the intestinal microbiome significantly decreased, and these trends were not correlated with neoadjuvant chemotherapy efficacy. Fusobacterium abundance remained significantly higher in poor responders than good responders post-NAC, thus suggesting its association with chemoresistance. The Firmicutes/Bacteroidetes ratio was lower in patients with breast cancer than healthy controls, and was correlated with the therapeutic response: this ratio rose post-NAC but remained suboptimal in poor responders. Untargeted metabolomics identified upregulated amino acids (Thr-Thr and histidine) in poor responders and elevated lipids (C17-sphinganine) in good responders. ROC (receiver operating characteristic curve) analysis validated these metabolites (AUC >0.7) as predictive biomarkers. KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis highlighted enrichment in mTOR signaling, endocrine resistance, and estrogen signaling pathways.
Conclusions: These findings underscore the intestinal microbiome’s potential as a predictor of NAC efficacy and a therapeutic target. Modulating Fusobacterium or metabolite pathways may enhance chemotherapy response.
Keywords
Breast cancer, intestinal microbiome, metabolites, neoadjuvant chemotherapy.
Introduction
Breast cancer remains one of the most prevalent malignancies among women worldwide, and its incidence has steadily increased in recent years [1]. Current therapeutic strategies include surgery, endocrine therapy, immunotherapy, chemotherapy, and radiation [2]. Chemotherapy for breast cancer is divided into preoperative and postoperative chemotherapy. Preoperatively administered neoadjuvant chemotherapy, a standard approach for locally advanced breast cancer, not only improves surgical feasibility but also guides subsequent treatment regimens according to therapeutic response [3]. The Miller-Payne (MP) grading system, a pathological evaluation method for assessing decreased tumor cellularity post-treatment, is widely used in China to predict patient prognosis [4]. Regular monitoring of neoadjuvant chemotherapy efficacy through clinical evaluations—such as physical examination, ultrasound, and magnetic resonance imaging—is critical for optimizing treatment decisions [5].
The intestinal microbiota, a symbiotic microbial community within the gastrointestinal tract, plays critical roles in host physiology, including metabolism, immune regulation, and disease progression; moreover, a symbiotic relationship is believed to exist between humans and their intestinal microbiota [6–8]. Dysbiosis, an imbalance in microbial composition, can perturb the host-microbiota relationship and drive tumor development. Emerging evidence has linked intestinal dysbiosis (microbial imbalance) to carcinogenesis and treatment outcomes across multiple cancer types [9–11], including stomach, colon, liver, lung, skin, and breast cancers [12–17]. Notably, patients with breast cancer exhibit intestinal microbial profiles that are distinct from those of healthy individuals, and are characterized by altered bacterial diversity, relative abundance, and functional pathways [17–19].
The intestinal microbiota can transform host nutrients into complex metabolites [20]. Furthermore, intestinal microbiota-derived metabolites, such as short-chain fatty acids and bile acids, modulate host signaling pathways, thereby influencing tumor progression and drug metabolism. Recent studies have suggested that chemotherapy agents, including anthracyclines and taxanes, interact bidirectionally with the intestinal microbiota: chemotherapy alters microbial composition, whereas the microbiota metabolizes drugs, thereby affecting their efficacy and toxicity [21–24]. Microbial abundance and diversity have been reported to significantly decrease after chemotherapy [25, 26]. Moreover, the intestinal microbiota and its relevant metabolites may play important roles in both the progression and treatment of breast cancer. Monitoring neoadjuvant chemotherapy efficacy is essential for patients with breast cancer. Therefore, on the basis of previous studies, we hypothesized that monitoring neoadjuvant chemotherapy efficacy for breast cancer according to changes in the intestinal microbiota and relevant metabolites might provide a powerful tool. This study investigated the interplay among intestinal microbial shifts, metabolite profiles, and neoadjuvant chemotherapy efficacy in patients with breast cancer, to identify potential biomarkers for therapeutic optimization.
Methods and materials
Sample collection and preservation
From May 2021 to December 2021, 16 patients diagnosed with breast cancer through needle biopsy who received neoadjuvant chemotherapy before undergoing surgery at the Department of Breast Surgery of the First Affiliated Hospital of Nanjing Medical University were included in this study.
Inclusion criteria
- Patients 18–60 years of age, with puncture pathology indicating an invasive breast cancer diagnosis, who met any of the following:
- Tumor ≥5 cm
- Tumor invasion of the skin or chest wall
- Clinical evaluation of axillary lymph node metastasis cN2 or above
- Neoadjuvant chemotherapy regimen of EC-T for a total of eight cycles. First four cycles: epirubicin 90 mg/m2, cyclophosphamide 600 mg/m2, once every 3 weeks. Last 4 cycles: docetaxel 80 mg/m2, once every 3 weeks. HER-2 positive patients with breast cancer were treated with herceptin targeted therapy in the last four cycles of chemotherapy, with an initial dose of 8 mg/kg and a maintenance dose of 6 mg/kg. After eight cycles of neoadjuvant chemotherapy treatment, all patients underwent surgical treatment.
- Normal body mass index (BMI), 16–26 kg/m2
- To minimize confounding effects from dietary variations, all participants received a standardized, nutritionally complete, and hygienically prepared diet throughout the neoadjuvant chemotherapy period. This controlled diet protocol ensured adequate caloric intake with a balanced ratio of high-quality animal protein (e.g., lean poultry, fish, eggs) and plant-based protein (e.g., legumes, tofu). Additionally, it provided sufficient essential vitamins, minerals, and dietary fiber and excluded known prebiotic and probiotic supplements or fermented foods.
Exclusion criteria
- History of malignant tumors or receipt of anti-tumor drug treatment
- Complications of severe cardiopulmonary disease, or liver or kidney dysfunction
- Acute or chronic inflammatory bowel disease
- Receipt of antibiotics, intestinal probiotics, gastrointestinal motility drugs, or other drugs affecting the intestinal flora within 4 weeks before fecal collection
- Failure to complete standard course of treatment
According to the above criteria, three patients were excluded. Fecal specimens were collected from patients with a sterilized collection kit and immediately stored at −80°C until further analysis. After all samples were collected, extraction and sequencing were performed for each batch.
DNA extraction and PCR amplification
According to the manufacturer’s instructions, a PowerSoil DNA Isolation Kit (MO BIO Laboratories, Carlsbad, CA, United States) was used to extract DNA from stool samples. The purity and quality of genomic DNA were verified on 1% agarose gels and with a NanoDrop spectrophotometer (Thermo Scientific).
The hypervariable region of V3–V4 16S rRNA was amplified by PCR with the primers 338F (5′-ACTCCTACGGGAGGCAGCAG-3′) and 806R (5′-GGACTACNNGGGTATCTAAT-3′) [27]. For each stool sample, the amplification conditions were as follows: 95°C for 5 min, followed by 30 cycles of 94°C for 30 s, 50°C for 30 s, and 72°C for 60 s, and a final extension at 72°C for 10 min. The PCR products were purified with an Agencourt AMPure XP Kit.
High-throughput sequencing
High-quality PCR products were sequenced on a MiSeq PE300 high-throughput sequencing system (Illumina, San Diego, CA, United States), and technical support was provided by Beijing Allwegene Technology (Beijing, China). After the run, image analysis, base calling, and error estimation were performed with Illumina Analysis Pipeline version 2.6.
Original data processing
Quality control processing of the raw data included removal of sequences that were shorter than 230 bp or had low-quality scores (≤20) with the UPARSEOTU algorithm (v2.7.1; USEARCH software, https://drive5.com/uparse), and deletion of chimeric sequences with the UCHIME algorithm (USEARCH; https://www.drive5.com/usearch/manual/uchime_algo.html) based on the “Gold database.” Clean tags were clustered into operational taxonomic units (OTUs) at a similarity level of 97% with the Uparse algorithm in Vsearch (v2.7.1) software [28].
QIIME (v1.8.0; http://qiime.org/1.8.0) software was used to generate rarefaction curves and to analyze the richness and diversity indices according to the OTU information. Smooth curves suggested that the produced data were sufficient.
Alpha diversity
Alpha diversity was used to analyze the complexity of species diversity in QIIME software (v1.80) for each sample, on the basis of the Chao1, Observed species, Shannon, and Simpson indices [29], all of which positively correlate with sample complexity. Chao1 value and Observed species values reflect the species richness of the community. The Shannon value and Simpson value reflect the species diversity of the community, and are influenced by species richness and evenness; that is, both values consider the abundance of each species. With the same species richness, greater species evenness is associated with greater community diversity.
Beta diversity analysis
Beta diversity analysis was used to evaluate differences in fecal samples in bacterial community structure in QIIME software (v1.80). Beta diversity was measured with both weighted and unweighted UniFrac, Bray-Curtis, and principal coordinate analysis (PCOA). The differences between two communities are commonly reflected by the Bray-Curtis distance and visualized with PCOA, whereas UniFrac uses the system evolution information to compare the community species composition between samples. These results consider the distance of evolution between species, and higher index values are associated with greater differences between samples. Therefore, the results can be used to measure beta diversity.
Untargeted analysis of fecal metabolites
Fecal samples were added to extraction solvent, and the extract was diluted after grinding, ultrasonic processing, and centrifugation. Supernatants were further separated with an Agilent 1290 Infinity II series UHPLC System (Agilent Technologies, Santa Clara, CA, United States) equipped with a Waters ACQUITY UPLC BEH Amide column (2.1 × 100 mm, 1.7 μm). Subsequently, mass spectrometry data were obtained with a Triple TOF 6600 mass spectrometer (AB SCIEX, Redwood City, CA, United States). The raw data were converted into mzML format with ProteoWizard and preprocessed with the R package XCMS v3.2.78. The processed data included the peak intensity, mass-to-charge ratio, and retention time. Data management was conducted as follows: peaks in less than 50% of quality control (QC) samples or 80% of biological samples were removed. Features with relative standard deviation >30% were excluded. Subsequently, the differential metabolites were filtered through statistical analysis. The significant metabolites had variable importance in projection (VIP) ≥1 and P value (T test) <0.05. We further identified significant metabolites on the basis of >1.25-fold change for subsequent analysis. The metabolite pathways were searched with databases including KEGG (http://www.kegg.jp), HMDB (http://www.hmdb.ca), and MetaboAnalyst (http://www.metaboanalyst.ca)
Statistical analysis
Statistical analysis and plotting were performed in R (version 3.6.0), SPSS 25.0, and GraphPad Prism 9.0 software. Fisher‘s exact test was used for count data. The normality of data distribution was tested with single factor ANOVA. Non-normally distributed data were analyzed with the Wilcoxon rank sum test. Differences between groups were analyzed with the Kruskal-Wallis test. Four groups of weighted and unweighted UniFrac distances were compared through molecular analysis of variance (AMOVA). The receiver operating characteristic curve (ROC) method was used to predict efficacy. P < 0.05 indicated statistical differences.
Results
Intestinal microbiome dysbiosis is closely associated with neoadjuvant chemotherapy efficacy in breast cancer
Characteristics of study participants
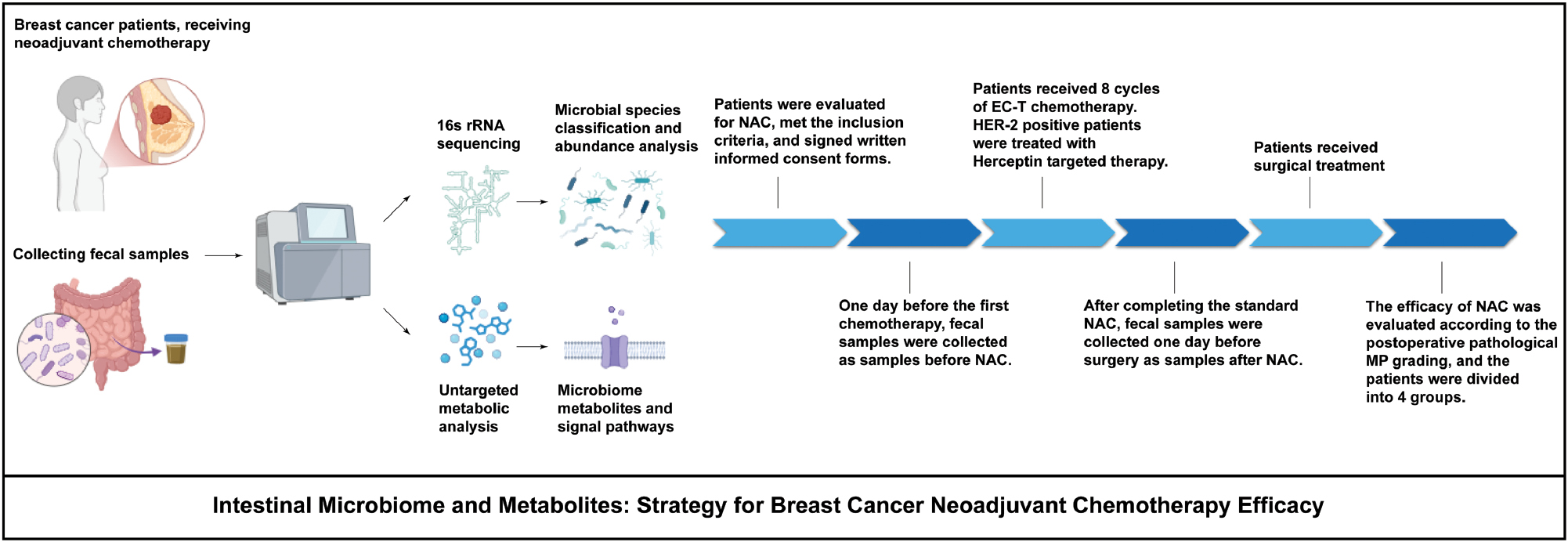
A total of 26 fecal samples from 13 patients with breast cancer were included in this study (patient clinical characteristics in Table 1). The standard treatment process (including neoadjuvant chemotherapy and surgical treatment), sample collection times, processing, and analysis after sample collection are shown in Figure 1. Patients with breast cancer were divided into four groups according to neoadjuvant chemotherapy efficacy, according to the MP grading system. Stool samples of patients with a good chemotherapy response (MP grades 4–5) were divided into groups a and b, representing those before receipt of the first treatment and after the last treatment, respectively. Stool samples of patients with poor response (MP grades 1–3) were divided into groups c and d, representing those before receipt of the first treatment and after the last treatment, respectively.
Table 1 Clinical Characteristics of Patients by MP Grade
| Category | Total | MP grade low remission | MP grade high remission | P value | |
|---|---|---|---|---|---|
| Total | 13 | 6 | 7 | ||
| Age | <50 | 7 | 3 | 4 | 1.000 |
| ≥50 | 6 | 3 | 3 | ||
| ER | Negative | 5 | 3 | 2 | 0.592 |
| Positive | 8 | 3 | 5 | ||
| PgR | Negative | 6 | 3 | 3 | 1.000 |
| Positive | 7 | 3 | 4 | ||
| HER-2 | Negative | 5 | 4 | 1 | 0.102 |
| Positive | 8 | 2 | 6 | ||
| Ki-67 before NAC | <50% | 5 | 3 | 2 | 0.592 |
| ≥50% | 8 | 3 | 5 | ||
| Ki-67 after NAC | <50% | 8 | 2 | 6 | 0.102 |
| ≥50% | 5 | 4 | 1 | ||
| Lymphatic metastasis | Without metastasis | 6 | 2 | 4 | 0.592 |
| Without metastasis | 7 | 4 | 3 | 0.940 | |
| Histologic grade | 2 | 5 | 2 | 3 | |
| 3 | 6 | 3 | 3 | ||
| Unable to assess | 2 | 1 | 1 |
Figure 1 The standard treatment process, and sample collection, preparation, and analysis, in patients with breast cancer receiving neoadjuvant chemotherapy.
OTU analysis
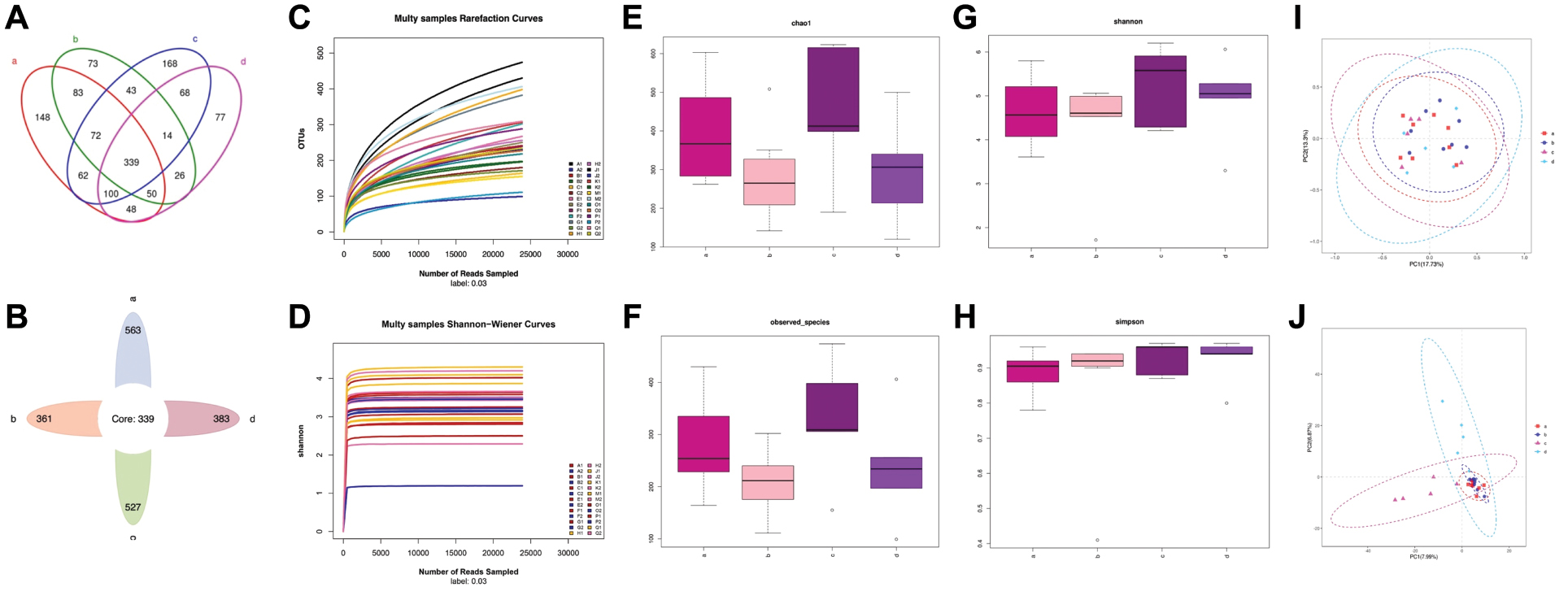
OTU clustering was performed for the sequences at 97% similarity, and bioinformatics statistical analysis was subsequently conducted. A Venn diagram of OTUs indicated 902 OTUs in group a, 866 OTUs in group c, and 573 shared OTUs. In contrast, group b comprised 700 OTUs, group d comprised 722 OTUs, and the two groups shared 429 OTUs. The non-overlapping portions represented the number of unique OTUs in each group. The number of OTUs shared by all groups was 339 (Figure 2A). The intestinal microbiota OTUs in groups a and c were all higher than those in groups b and d (Figure 2B). The number of OTUs decreased after neoadjuvant chemotherapy, regardless of efficacy.
Figure 2 (A) Venn diagram of OTUs. (B) Flower diagram of OTUs. (C) Dilution curve of the alpha diversity index. The dilution curve (Sobs index) tends to be gentle. (D) Shannon-Wiener curves tended to flatten, thus indicating relatively stable intestinal microbiota in all patients. (E)–(H) Alpha diversity index boxplot for the four groups, according to the Chao1, observed species, Shannon, and Simpson indices. Abnormal values are shown as “o.” (I) Principal coordinate analysis score plots for all groups. Groups are presented in different colors (group a, red; group b, blue; group c, light purple; and group c, light blue). The closeness among the four groups indicates similar species composition among the four samples. (J) PLS-DA score plot of species abundance samples from participants in the four groups.
Alpha diversity analysis
The flattening of the dilution curve of the alpha diversity index curve indicated sufficient sample sequencing and sequencing depth (Figure 2C). That is, few undiscovered species were present among the detected species, thus ensuring the reliability of our study.
The microbial species abundance (quantity of different species) and evenness (quality of being balanced in the environment) are frequently measured with the Shannon diversity index. The Shannon diversity index of intestinal microbiota was comparatively stable in all patients (Figure 2D).
To characterize the fecal microbiome community diversity (richness and evenness) in the four groups, we calculated the Chao1, observed species, Shannon, and Simpson indexes to estimate the alpha diversity. Significant differences in overall bacterial community structure across the four groups were found in the Chao1 and observed species indices (Figure 2E and 2F). Moreover, the microbial communities in groups a and c had greater species richness than those in groups b and d. Therefore, patients with good or poor chemotherapy response had higher fecal microbiome community diversity before rather than after chemotherapy. No significant differences were observed in the diversity of communities (species richness and evenness) across the four groups, according to the Shannon and Simpson indices (Figure 2G and 2H), thus indicating similar community composition patterns among the four groups. However, the value of the Simpson index approached 1, thus suggesting that microbial communities of the four groups had high species-level diversity. This finding implied that as the species richness and evenness increased, the diversity increased.
Beta diversity analysis
To visualize the overall differences in beta diversity in species among the microbiome profiles of the four groups, we conducted PCOA of weighted and unweighted UniFrac distances. The closeness of the four samples indicated similar species composition. The generated weighted and unweighted UniFrac distance metrics did not indicate significant differences in beta diversity between the groups with good or poor response to neoadjuvant chemotherapy, both before or after therapy (Figure 2I).
Partial least squares discriminant analysis (PLS-DA) regression models are often used to predict the clinical status of samples. We used PLS-DA to compare the intestinal microbial profiles among the four groups. The results indicated a distinct clustering pattern among samples before and after neoadjuvant chemotherapy in patients with breast cancer, regardless of whether the efficacy of therapy was good or poor (Figure 2J), thus reflecting the separation in microbial composition.
Structural analysis of microbiota associated with breast cancer
Microbial composition and changes before and after neoadjuvant chemotherapy in the four groups at the phylum level
In our study cohort, among patients with poor neoadjuvant chemotherapy efficacy, the co-existing intestinal microbiota colonies at the phylum level included Firmicutes, Bacteroidetes, Fusobacteria, Proteobacteria, and Actinobacteria (specific relative abundance of individual specimens in Figure 3A). Among all patients with poor efficacy, Fusobacteria and Proteobacteria in the intestinal microbiota both decreased after neoadjuvant chemotherapy, with an average percentage decrease of 1.870% and 1.203%, respectively (Figure 3B).
Figure 3 Phylum level bacterial composition and changes before and after neoadjuvant chemotherapy in the four groups. (A) Pie chart of bacterial species and proportions in the four groups. (B) Bacteria with the same change trends in all samples of each group before and after neoadjuvant chemotherapy, in groups with poor or good neoadjuvant chemotherapy efficacy. The percentage is accurate to 2–3 decimal places.
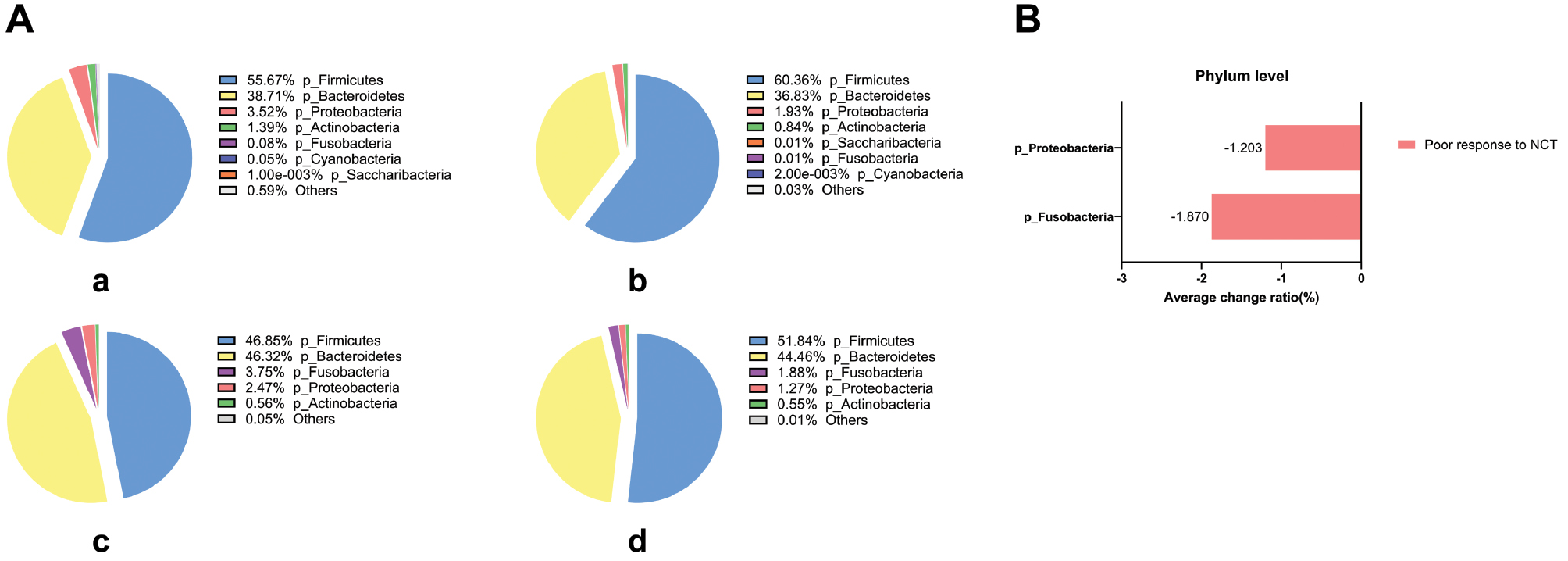
Among patients with good neoadjuvant chemotherapy efficacy, the co-existing bacterial colonies included Firmicutes, Bacteroidetes, Proteobacteria, Actinobacteria, Fusobacteria, Cyanobacteria, and Saccharibacteria (specific relative abundance of individual specimens in Figure 3A). However, among all patients with good efficacy, we did not confirm the same change trend in the intestinal microbiota before and after neoadjuvant chemotherapy (Figure 3B).
Microbial composition and changes before and after neoadjuvant chemotherapy in the four groups at the class, order, and family levels
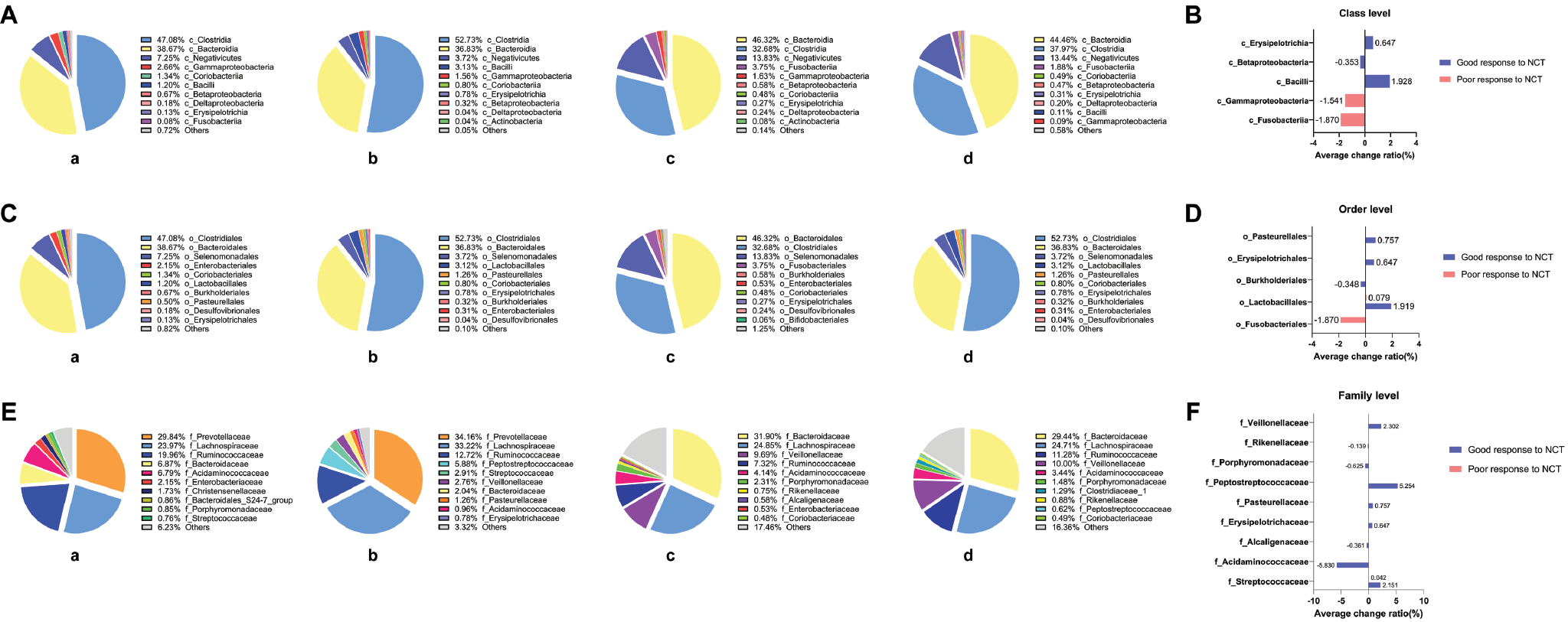
At the class, order, and family levels, among patients with poor or good neoadjuvant chemotherapy efficacy, the compositions of co-existing bacterial colonies and their average change percentages are shown in Figure 4. Notably, among all patients with poor efficacy, Fusobacteria decreased by an average of 1.870% at the class level, and Fusobacteriales decreased by 1.870% at the order level, after neoadjuvant chemotherapy. In addition, among patients with poor efficacy or good efficacy, the abundance of Lactobacillales at the order level and Streptococcaceae at the family level increased after neoadjuvant chemotherapy, but this increase was much clearer among the patients with good efficacy.
Figure 4 Main bacterial composition and changes before and after neoadjuvant chemotherapy in the four groups at the following levels: (A), (B) class level; (C), (D) order level; (E), (F) family level.
Microbial composition and changes before and after neoadjuvant chemotherapy in the four groups at the genus level
At the genus level, among patients with poor neoadjuvant chemotherapy efficacy, the co-existing intestinal microbiota included 52 bacterial colonies, among which Bacteroidaceae, Lachnospiraceae, Veillonellaceae, Ruminococcaceae, Acidaminococcaceae, and Porphyromonadaceae were the most important. The top ten types of bacteria at the order level and their relative abundance in individual specimens are shown in Figure 5A. Among all patients with poor efficacy, Parabacteroides decreased after neoadjuvant chemotherapy, by an average of 1.076%. We observed increased abundance of seven types of bacteria, including the Eubacterium rectale group (5.179% average increase), Butyricicoccus (0.611%), Lachnospiraceae ND3007 group (0.567%), Eubacterium hallii group (0.322%), Dorea (0.140%), Streptococcus (0.044%), and Veillonella (0.023%) (Figure 5B).
Figure 5 Main bacterial composition and changes before and after neoadjuvant chemotherapy in the four groups at the genus level. (A) Pie chart of the top ten most abundant bacterial species and proportions for the four groups. (B) Bacteria with the same change trend in all samples in each group before and after neoadjuvant chemotherapy, among groups with poor or good neoadjuvant chemotherapy efficacy. The percentage is accurate to 2–3 decimal places.
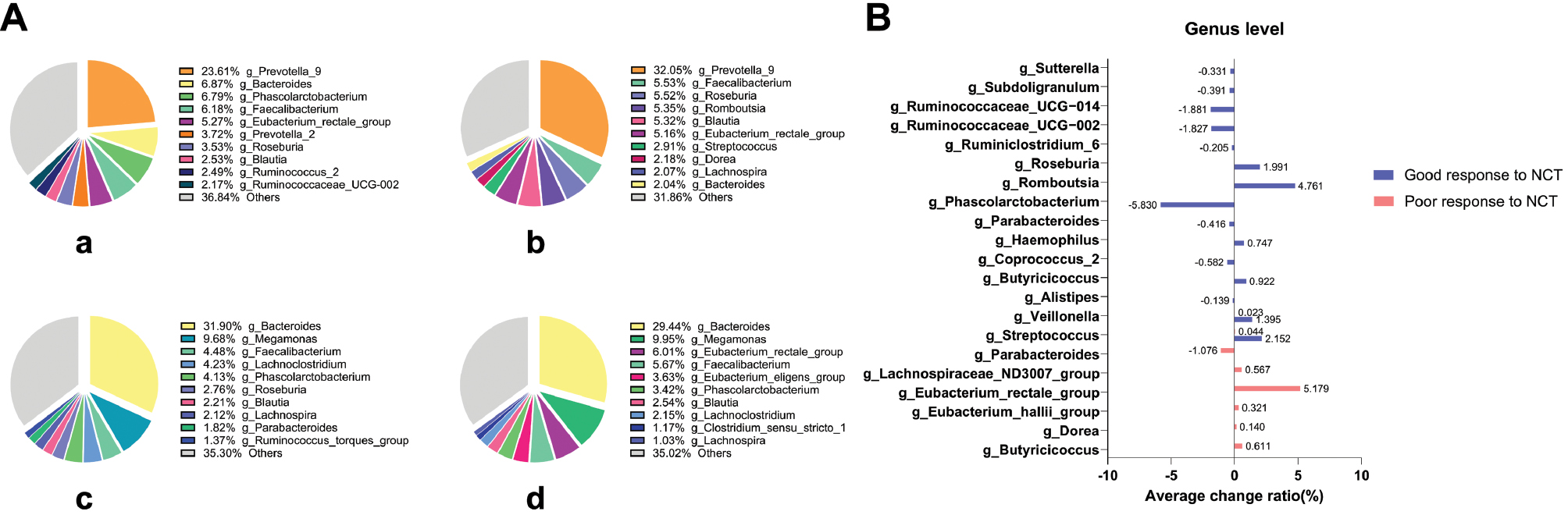
Among patients with good neoadjuvant chemotherapy efficacy, the co-existing intestinal microbiota included 78 bacterial colonies, among which Prevotella_9, Bacteroides, Faecalibacterium, Eubacterium rectale group, Prevotella_2, Roseburia, Blautia, Lachnospira, and Coprococcus_2 were the most important. The top ten types of bacteria at the order level and their relative abundance in individual specimens are shown in Figure 5A. Among all patients with good efficacy, a total of 15 bacterial colonies showed the same change trend (nine decreasing and six increasing) before and after neoadjuvant chemotherapy. In detail, the bacterial colonies with decreased abundance after neoadjuvant chemotherapy included Phascolarctobacterium (5.829% average decrease), Ruminococcaceae_UCG-014 (1.881%), Ruminococcaceae_UCG-002 (1.827%), Coprococcus_2 (0.582%), Parabacteroides (0.416%), Subdoligranulum (0.390%), Sutterella (0.331%), Ruminiclostridium_6 (0.205%), and Alistipes (0.139%). The colonies with increased abundance included Romboutsia (4.761% average increase), Streptococcus (2.152%), Roseburia (1.991%), Veillonella (1.395%), Butyricicoccus (0.922%), and Haemophilus (0.747%) (Figure 5B).
Intestinal microbiota composition or changes potentially associated with neoadjuvant chemotherapy efficacy
Notably, at the phylum, class, and order levels, Fusobacteria, Fusobacteria, and Fusobacteriales had higher abundance among patients with poor rather than good neoadjuvant chemotherapy efficacy. At the family and genus levels, Fusobacteriaceae was found in most patients with poor neoadjuvant chemotherapy efficacy, and showed high abundance and substantial changes before versus after neoadjuvant chemotherapy in these patient samples but not all patient samples. Therefore, we analyzed the abundance and changes in Fusobacterium at various levels in all patient samples except for one in which the abundance of Fusobacteriaceae (at the family level) and Fusobacterium (at the genus level) could not be detected. As shown in Figure 6A and 6B, among patients with poor neoadjuvant chemotherapy efficacy, the baseline abundance of Fusobacterium before chemotherapy was significantly higher than observed in patients with good efficacy. After neoadjuvant chemotherapy, although the abundance of Fusobacterium in both groups showed a decreasing trend, the abundance of Fusobacterium in patients with poor efficacy was still much higher than the baseline level in patients with good efficacy, thus suggesting that high abundance of Fusobacterium in vivo might be associated with poor neoadjuvant chemotherapy efficacy. Consequently, to increase neoadjuvant chemotherapy efficacy, the abundance of Fusobacterium might need to be decreased below a certain level.
Figure 6 (A) Average abundance of Fusobacterium at different levels before and after neoadjuvant chemotherapy of the four groups. (B) Average change ratio of Fusobacterium at different levels. (C) Specific composition of Firmicutes and Bacteroidetes at the phylum level of the four groups. (D) Firmicutes/Bacteroidetes (F/B) ratio of the four groups.

The Firmicutes/Bacteroidetes (F/B) ratio in patients with breast cancer was lower than that in healthy controls. The ratio was approximately 6 in healthy individuals and 2 in patients with breast cancer, thus indicating that the F/B ratio might be used as a breast cancer risk factor and might potentially explain the mechanisms affecting breast cancer development [30]. Similarly, the baseline F/B ratio was higher among patients with good rather than poor efficacy. After neoadjuvant chemotherapy, the F/B ratio in both groups increased; however, among patients with poor efficacy, the F/B ratio after treatment was close to the baseline level observed in patients of good efficacy. Moreover, the F/B ratio was usually below 2 in patients with breast cancer regardless of neoadjuvant chemotherapy receipt or efficacy (Figure 6C and 6D). Therefore, drugs targeting specific intestinal microbiota aimed at increasing the F/B ratio might potentially increase neoadjuvant chemotherapy efficacy; however, more samples must be analyzed to provide verification of this possibility.
In addition, among patients with good response, the increase in Streptococcaceae and Veillonellaceae (at the family level), and Streptococcus and Veillonella (at the genus level), might be associated with good efficacy.
Changes in fecal microbial metabolites after neoadjuvant chemotherapy
To determine whether microbiome metabolic pathways might influence the metabolism of the efficacy of the neoadjuvant chemotherapy, we conducted untargeted metabolic analysis of fecal samples from patients with breast cancer who received neoadjuvant chemotherapy. A total of 1296 metabolites were detected after data management. An OPLS-DA model was established between the good-response and poor-response to chemotherapy groups and shows good discrimination. The OPLS-DA score plots indicated that the samples formed two clusters with clear separation, this further confirming significant metabonomic differences between groups (Figure 7A). On the basis of the S-plot from OPLS-DA analysis, 472 metabolites with a VIP score >1 were selected (Figure 7B). Among them, 28 differential metabolites were identified according to the selection criteria, including 12 significantly upregulated and 16 downregulated metabolites (Figure 7C). In general, these compounds comprised carbohydrates, lipids, amino acids, steroids, and their derivatives. Notably, most of the lipids and lipid-like molecules were significantly diminished, whereas the amino acids and carbohydrates were elevated. The fold differences in differential metabolites were visualized in a histogram (Figure 7D), in which red represents up-regulated metabolites, and green represents down-regulated metabolites.
Figure 7 (A) OPLS-DA plots with scores for the two groups (a_c, patients with good neoadjuvant chemotherapy efficacy; b_d, patients with poor neoadjuvant chemotherapy efficacy). (B) S-plots of the OPLS-DA model for the two groups. (C) Volcano plot of differential metabolites obtained according to the screening criteria from all metabolites (p value <0.05, VIP ≥1, and fold change <0.67 or >1.5). (D) Histogram of differential metabolites, visualized as fold difference. (E) Variable importance in projection (VIP) plot obtained from OPLS-DA.
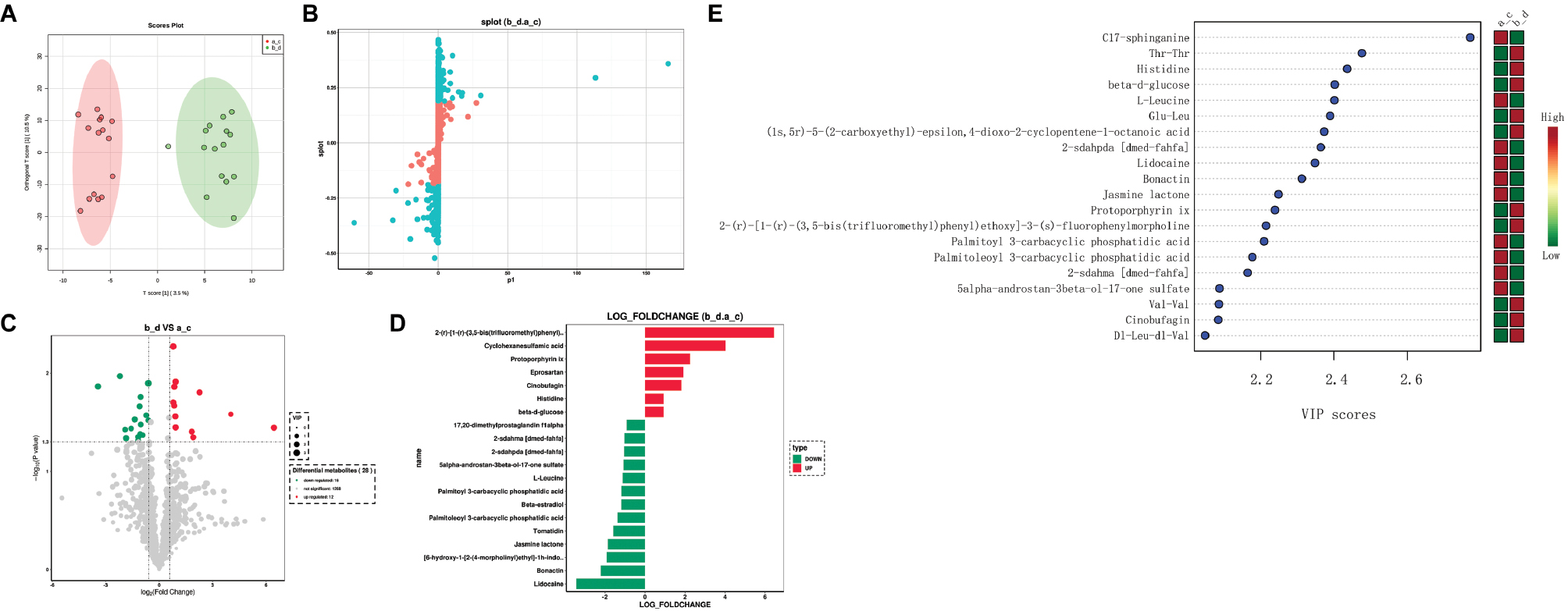
We analyzed differential metabolites between the a_c (patients with good neoadjuvant chemotherapy efficacy) and b_d (patients with poor neoadjuvant chemotherapy efficacy) groups (top 20 most relevant metabolites in Figure 7E). We used the top three ranked differential metabolites to explore the potential key metabolites in the two groups, according to the VIP plot obtained from OPLS-DA. Among them, C17-sphinganine was indicative of good neoadjuvant chemotherapy response, whereas Thr-Thr and histidine were indicative of poor neoadjuvant chemotherapy response. In addition, to illustrate the potential diagnostic value of the intestinal microbiome metabolism in predicting neoadjuvant chemotherapy efficacy in breast cancer, we developed a random forest model based on the differential metabolites. Univariate ROC analysis revealed that 20 of 28 differential metabolites had AUC values exceeding 0.7. The ROC curves of three representative metabolites (C17-sphinganine, Thr-Thr, and histidine) are shown in Figure 8A, 8B, and 8C. The results showed high discriminatory power in predicting neoadjuvant chemotherapy efficacy.
Figure 8 (A)–(C) ROC analysis, indicating good diagnostic value of the three representative metabolites in terms of neoadjuvant efficacy. (D) Bubble plot of the KEGG pathway enrichment analysis of markedly differential metabolites.
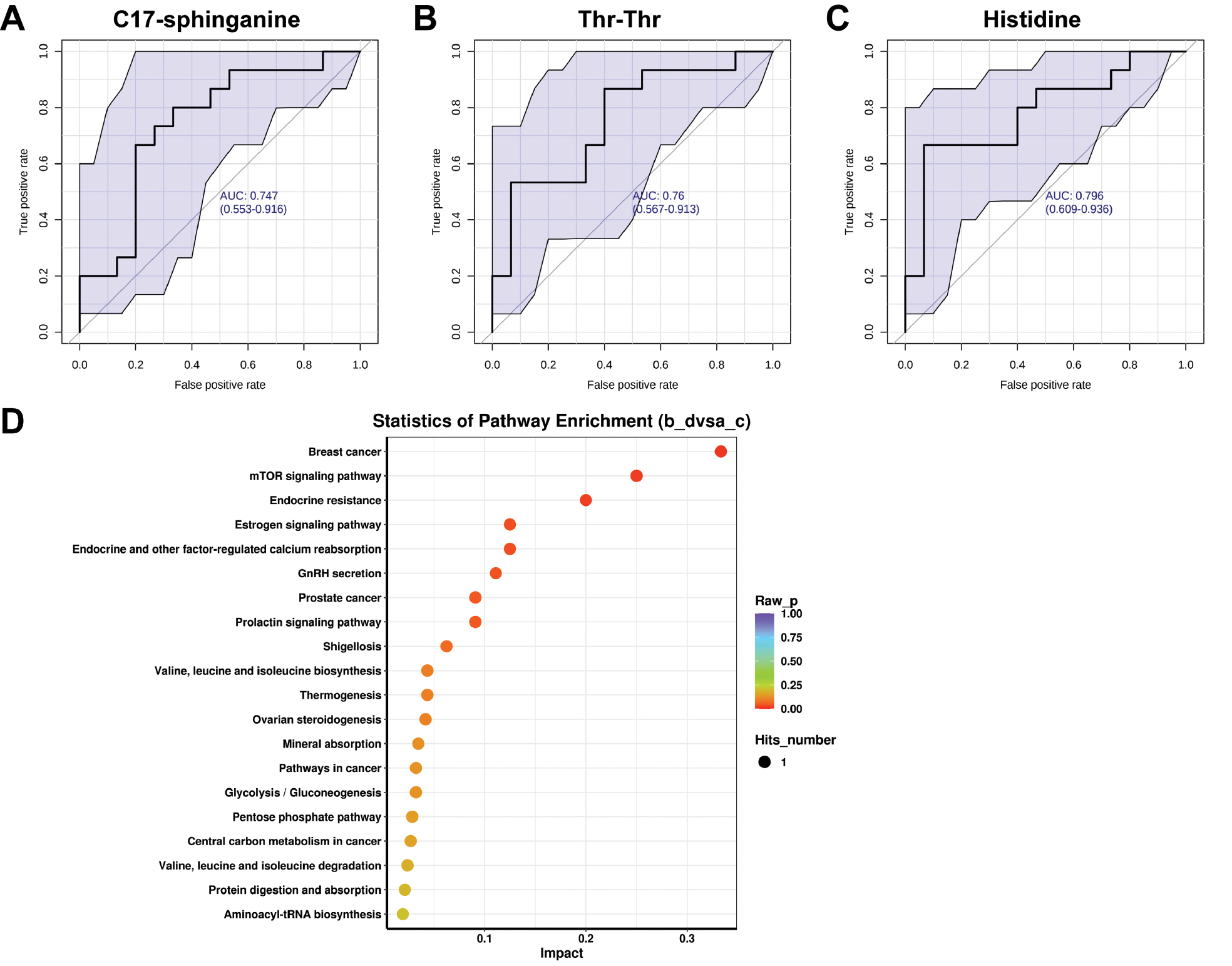
KEGG pathway differential enrichment analysis of the metabolites was conducted to identify the top 20 pathways according to P values (Figure 8D). The targets of different clusters were distributed primarily in pathways associated with breast cancer, and related genes were enriched in breast cancer-associated pathways, e.g., the mTOR signaling pathway, endocrine resistance, and estrogen signaling pathway.
Discussion
Microbiota and their relevant metabolites have been a focus of numerous research investigations in recent years. Bacteria in the intestinal microbiota influence chemotherapy in cancers including breast cancer [31]. Among the various neoadjuvant chemotherapy regimens for breast cancer, the combination of anthracyclines (epirubicin or doxorubicin), alkylating agents (cyclophosphamide), and taxanes (docetaxel or paclitaxel) is widely used [32]. Anthracycline drugs can regulate the relative species abundance and content of the intestinal flora, and several specific bacteria metabolize anthracycline drugs [33–35]. The use of cyclophosphamide damages the intestinal mucosa, and the stability of the intestinal flora can prevent mucosal damage caused by cyclophosphamide [36]. Taxanes may also undergo bacterial metabolism and interfere with the composition of the intestinal flora [37]. This study focused on the changes in intestinal flora and metabolites in patients with breast cancer who were administered epirubicin, cyclophosphamide, and docetaxel as neoadjuvant chemotherapy drugs. A comprehensive analysis of these changes before and after chemotherapy was conducted. Our results clarified the composition characteristics and related changes in the gut microbiota before and after neoadjuvant chemotherapy. These changes significantly differed between high-remission and low-remission patients.
Our results confirmed a decline in intestinal microbiota diversity at the phylum, class, order, family, and genus levels after neoadjuvant chemotherapy, in patients with good or poor efficacy. Compared with patients with good efficacy, patients with poor efficacy had a lower number of intestinal microbiome species before NAC, and the reduction of flora species after NAC was more significant. In addition, among patients with good neoadjuvant chemotherapy efficacy, the intestinal microbiota composition and variation trends compared with patients with poor efficacy, the composition and change trend of intestinal microbiome before and after NAC in patients with good efficacy were more consistent. Moreover, the abundance of specific bacterial groups confirmed to be associated with breast cancer in prior studies [38, 39], such as Firmicutes, Clostridia, Proteobacteria, and Bacteroidota, all showed changes in their relative proportions in our study. These changes therefore might contribute to the efficacy of breast cancer treatment. In general, to increase the accuracy of the conclusions, larger numbers of patient samples should undergo further detection and verification. We also observed some differences in the significance of related changes at different levels; consequently, combinations of multiple markers must be examined to improve accuracy and sensitivity in clinical diagnosis and treatment. Herein, we focused on the flora showing consistent trends at various levels.
Fusobacterium, represented by Fusobacterium nucleatum, is an oral pathogen that is frequently translocated and engrafted in the upper gastrointestinal tract. Increased abundance of Fusobacterium is frequently associated with colorectal cancer [40]. The mechanisms through which Fusobacterium promotes tumorigenesis include establishing an immunosuppressive tumor microenvironment, affecting T cell infiltration and macrophage polarization [41], promoting tumor cell proliferation and immune escape, inducing chemotherapy resistance, and activating immune checkpoints. In addition, Fusobacterium can transfer to breast tissue through the mammary-intestinal axis, direct nipple contact, or hematogenous transmission [42, 43]. Mechanistically, Fusobacterium may activate the Toll-like receptor 4 (TLR4) pathway [44, 45] and induce up-regulated expression of matrix metalloproteinase-9 (MMP-9) [42], thereby promoting the formation of an immunosuppressive and tumor-promoting microenvironment. Simultaneously, the high expression of immune checkpoint receptors on T cells, natural killer cells, and tumor-infiltrating lymphocytes induced by Fusobacterium is also involved in tumor immune evasion [42]. However, Fusobacterium increases the expression of immune checkpoints, thus potentially increasing immune checkpoint therapy efficacy [46, 47]. In chemotherapy, Fusobacterium might interact with specific drugs and activate related pathways, thereby leading to drug resistance or immune escape of tumor cells, and ultimately affecting the efficacy of chemotherapy [48]. In esophageal squamous cell carcinoma, Fusobacterium regulates the expression of endogenous LC3 and ATG7 and the formation of autophagosomes, thereby inducing resistance to 5-FU, CDDP, and docetaxel [49]. In colorectal cancer, Fusobacterium promotes cisplatin resistance through miR-135b and the TCF4/β-catenin complex [50]. Therefore, we plan to further explore the specific mechanism through which Fusobacterium leads to neoadjuvant chemotherapy resistance in breast cancer, to provide theoretical support for improving neoadjuvant chemotherapy regimens, developing antibacterial drugs or vaccines, and developing combination treatments with immune checkpoint therapy in the future.
During recent years, in an emerging research area, intestinal microbial metabolites have shown promising anticancer potential. Intestinal metabolites are synthesized in the microbiome and subsequently exert biological effects [51]. To investigate the interactions between the intestinal microbiome and metabolites, and their effects on the efficacy of the neoadjuvant chemotherapy, we evaluated the microbial diversity and abundance of metabolites in fecal samples and their association with neoadjuvant chemotherapy for breast cancer. The top three enriched pathways of differential metabolites were the mTOR signaling pathway, endocrine resistance, and estrogen signaling pathway, which are associated with breast cancer. The mTOR signaling pathway plays important roles in the differentiation, proliferation, apoptosis, and energetic and glucose metabolism in breast cancer cells, and consequently affects breast cancer development and prognosis [52, 53]. Estrogen receptor-positive (ER+) breast cancer is the most common breast cancer subtype. Endocrine therapy is an important treatment for ER+ breast cancer. The endocrine resistance and estrogen signaling pathways play important roles in endocrine therapy, and are associated with drug resistance development and metabolic adaptation during breast cancer progression [54–57]. We further observed that most of the lipids and lipid-like molecules significantly decreased, whereas the amino acids and carbohydrates increased, after treatment. Among the three signature metabolites identified, C17-sphinganine, a carbohydrate, was indicative of patients who responded well to neoadjuvant chemotherapy, whereas the amino acids Thr-Thr and histidine were indicative of patients who responded poorly.
Soluble epoxide hydrolase (sEH) is involved in the inflammatory response through regulation of epoxyeicosatrienoic acids (EETs). sEH regulates the inflammatory response through the NF-κB and MAPK pathways, and regulates oxidative stress through the Nrf2 pathway [58, 59]. Imbalances in the intestinal flora imbalance (such as the excessive growth of pathogenic bacteria and a reduction of short-chain fatty acids) can induce intestinal mucosal inflammation and activate NF-κB and MAPK signaling pathways similar to sEH regulation. EETs, the substrates of sEH, have anti-inflammatory effects and are associated with intestinal barrier repair, through promoting the expression of tight junction proteins. sEH inhibitors increase the levels of EETs, which may directly regulate the composition of intestinal flora, such as by inhibiting pro-inflammatory bacteria and promoting probiotic colonization, or affecting flora metabolites through host metabolism, including that of short-chain fatty acids and bile acids [58–61]. Neoadjuvant chemotherapy for breast cancer (cisplatin and taxanes) often causes inflammation-associated adverse effects, such as mucositis and liver injury, and oxidative stress damage. Alisol B alleviates cisplatin-induced renal injury; therefore, similar mechanisms might be applicable to chemotherapy-associated organ toxicity [60]. The anti-inflammatory and antioxidant effects of sEH inhibitors might decrease chemotherapy damage to normal tissues. Inflammation in the tumor microenvironment (such as NF-κB pathway activation) is associated with chemotherapy resistance. sEH inhibitors inhibit macrophage/microglia activation and consequently might decrease the secretion of pro-inflammatory cytokines (TNF-α and IL-6), reverse the drug-resistant microenvironment, and enhance the efficacy of chemotherapy [61–63]. Studies have focused on the roles of sEH inhibitors in inflammation-associated diseases, such as acute lung injury, Parkinson’s disease, and acute kidney injury. The core mechanism involves decreasing inflammation and oxidative stress through regulation of EET levels and inhibiting NF-κB, MAPK, and other pathways. These targets (sEH) and pathways (inflammation and oxidative stress) are closely associated with the composition of intestinal flora and neoadjuvant chemotherapy in breast cancer. In addition, sEH inhibitors can inhibit macrophage activation, reduce the secretion of pro-inflammatory cytokines (TNF-α, IL-6), reverse the inflammatory microenvironment related to chemotherapy resistance, and therefore might have potential value in combined treatments. Future studies may explore the direct effects of sEH inhibitors on the composition of the intestinal flora, as well as the efficacy and mechanism in breast cancer models, and expand their clinical applications.
The sample size and research depth should be increased in future studies. This study was limited by the difficulty in obtaining clinical samples, and the time and cost of prospective studies. However, we controlled for confounding factors to the greatest extent possible, through strict inclusion and exclusion criteria and a paired design. Nonetheless, the small sample size might have led to bias in effect estimation and limited the external validity of our results. Notably, the flora-metabolite correlations observed herein showed stable trends; consequently, more robust conclusions would be expected to be obtained through expanding the sample size. Future research could build larger queues for verification through multi-center collaboration or integration of public database resources. At the level of mechanistic exploration, dozens of differential metabolites were successfully identified in this study, yet the exploration of their biological functions remains in its infancy. These metabolites might affect chemotherapy sensitivity by regulating the immune microenvironment and pharmacokinetics. Subsequent studies may include metabolic flow analysis, gene knockout models, and other technologies to systematically analyze the upstream and downstream regulatory networks of key metabolites, and elucidate their molecular mechanisms in the chemotherapy response. In addition, the interaction between microorganisms and metabolites has not been fully clarified. Fecal microbiota transplantation and sterile animal experiments are expected to further reveal the causal relationships between microbial metabolites and host chemotherapy responses. Nevertheless, this study reports what is, to our knowledge, the first combined analysis of microbial community and metabolites in the field of breast cancer chemotherapy efficacy prediction, thereby providing an innovative multi-omics perspective for exploring new cancer treatment targets. This study successfully revealed significant differences in intestinal flora structures and metabolite abundance between chemotherapy-sensitive and drug-resistant patients. These findings not only expand research frontiers in tumor treatment mechanisms but also offer potential biomarkers for the formulation of clinical personalized treatment strategies, thereby providing scientific value and clinical transformation potential.
Conclusion
Neoadjuvant chemotherapy alters the intestinal microbiota and relevant metabolites, and these changes might in turn influence chemotherapy efficacy. The intestinal microbiome and metabolite profiles undergo significant alterations during neoadjuvant chemotherapy that correlate with the therapeutic response. Targeting Fusobacterium or modulating metabolite pathways (e.g., C17-sphinganine, Thr-Thr, and histidine) might enhance chemotherapy efficacy. These findings underscore the intestinal microbiome’s potential as a predictive tool and therapeutic target in breast cancer management.
Data availability statement
The data used to support the findings of this study are included within the article.
Ethics statement
Written informed consent was obtained from all patients before enrollment in the study. The research proposal was approved by the institutional examination board of the First Affiliated Hospital of Nanjing Medical University (No. 2021-SR-476).
Author contributions
Tiansong Xia and Yichao Zhu conceptualized this study. Jingyue Fu collected clinical samples. Shuaikang Li and Yufan Jin conducted bioinformatics analysis. Hongxin Lin and Xingying Yu prepared the figures and tables. Jingyue Fu and Hongxin Lin wrote the draft. Hongxin Lin and Jie Mei revised the manuscript. Tiansong Xia and Yichao Zhu critically reviewed and edited the manuscript. Tiansong Xia obtained funding support. All authors read and approved the final manuscript.
Funding or acknowledgments
This study was supported by Jiangsu Province Hospital (2022-YJ-085-J-Z-ZZ-009).
Conflicts of interest
The authors declare that they have no conflicts of interest.
Graphical abstract
Highlights
The diversity of intestinal microbiome decreased after NAC, and the abundance of Fusobacterium was higher in drug-resistant patients. Metabolites (such as C17-sphinganine and Thr-Thr) can be used as predictive markers of efficacy.
In brief
In this study, 16S rRNA sequencing and metabolic analysis showed that the composition of the intestinal microbiome and metabolites was related to the efficacy of NAC in breast cancer, and Fusobacterium may be a drug resistance marker.
References
- Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, et al. Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2021;71(3):209-49. [PMID: 33538338 DOI: 10.3322/caac.21660]
- Trayes KP, Cokenakes SEH. Breast cancer treatment. Am Fam Physician 2021;104(2):171-8. [PMID: 34383430]
- Shien T, Iwata H. Adjuvant and neoadjuvant therapy for breast cancer. Jpn J Clin Oncol 2020;50(3):225-9. [PMID: 32147701 DOI: 10.1093/jjco/hyz213]
- Wang W, Liu Y, Zhang H, Zhang S, Duan X, et al. Prognostic value of residual cancer burden and Miller-Payne system after neoadjuvant chemotherapy for breast cancer. Gland Surg 2021;10(12):3211-21. [PMID: 35070881 DOI: 10.21037/gs-21-608]
- Wang H, Mao X. Evaluation of the efficacy of neoadjuvant chemotherapy for breast cancer. Drug Des Devel Ther 2020;14:2423-33. [PMID: 32606609 DOI: 10.2147/DDDT.S253961]
- Rossi P, Difrancia R, Quagliariello V, Savino E, Tralongo P, et al. B-glucans from Grifola frondosa and Ganoderma lucidum in breast cancer: an example of complementary and integrative medicine. Oncotarget 2018;9(37):24837-56. [PMID: 29872510 DOI: 10.18632/oncotarget.24984]
- Paul B, Barnes S, Demark-Wahnefried W, Morrow C, Salvador C, et al. Influences of diet and the gut microbiome on epigenetic modulation in cancer and other diseases. Clin Epigenetics 2015;7:112. [PMID: 26478753 DOI: 10.1186/s13148-015-0144-7]
- Jin C, Luo T, Zhu Z, Pan Z, Yang J, et al. Imazalil exposure induces gut microbiota dysbiosis and hepatic metabolism disorder in zebrafish. Comp Biochem Physiol C Toxicol Pharmacol 2017;202:85-93. [PMID: 28888875 DOI: 10.1016/j.cbpc.2017.08.007]
- McCoy KD, Mager LF. Impact of the microbiome on tumor immunity. Curr Opin Immunol 2021;69:39-46. [PMID: 33647829 DOI: 10.1016/j.coi.2021.01.002]
- Borella F, Carosso AR, Cosma S, Preti M, Collemi G, et al. Gut microbiota and gynecological cancers: a summary of pathogenetic mechanisms and future directions. ACS Infect Dis 2021;7(5):987-1009. [PMID: 33848139 DOI: 10.1021/acsinfecdis.0c00839]
- Park EM, Chelvanambi M, Bhutiani N, Kroemer G, Zitvogel L, et al. Targeting the gut and tumor microbiota in cancer. Nat Med 2022;28(4):690-703. [PMID: 35440726 DOI: 10.1038/s41591-022-01779-2]
- Mera RM, Bravo LE, Camargo MC, Bravo JC, Delgado AG, et al. Dynamics of Helicobacter pylori infection as a determinant of progression of gastric precancerous lesions: 16-year follow-up of an eradication trial. Gut 2018;67(7):1239-46. [PMID: 28647684 DOI: 10.1136/gutjnl-2016-311685]
- Gao R, Kong C, Huang L, Li H, Qu X, et al. Mucosa-associated microbiota signature in colorectal cancer. Eur J Clin Microbiol Infect Dis 2017;36(11):2073-83. [PMID: 28600626 DOI: 10.1007/s10096-017-3026-4]
- Yu LX, Schwabe RF. The gut microbiome and liver cancer: mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol 2017;14(9):527-39. [PMID: 28676707 DOI: 10.1038/nrgastro.2017.72]
- Lee SH, Sung JY, Yong D, Chun J, Kim SY, et al. Characterization of microbiome in bronchoalveolar lavage fluid of patients with lung cancer comparing with benign mass like lesions. Lung Cancer 2016;102:89-95. [PMID: 27987594 DOI: 10.1016/j.lungcan.2016.10.016]
- Yu Y, Champer J, Beynet D, Kim J, Friedman AJ. The role of the cutaneous microbiome in skin cancer: lessons learned from the gut. J Drugs Dermatol 2015;14(5):461-5. [PMID: 25942663]
- Plaza-Díaz J, Álvarez-Mercado AI, Ruiz-Marín CM, Reina-Pérez I, Pérez-Alonso AJ, et al. Association of breast and gut microbiota dysbiosis and the risk of breast cancer: a case-control clinical study. BMC Cancer 2019;19(1):495. [PMID: 31126257 DOI: 10.1186/s12885-019-5660-y]
- Chan AA, Bashir M, Rivas MN, Duvall K, Sieling PA, et al. Characterization of the microbiome of nipple aspirate fluid of breast cancer survivors. Sci Rep 2016;6:28061. [PMID: 27324944 DOI: 10.1038/srep28061]
- Xuan C, Shamonki JM, Chung A, Dinome ML, Chung M, et al. Microbial dysbiosis is associated with human breast cancer. PLoS One 2014;9(1):e83744. [PMID: 24421902 DOI: 10.1371/journal.pone.0083744]
- Anand S, Mande SS. Diet, microbiota and gut-lung connection. Front Microbiol 2018;9:2147. [PMID: 30283410 DOI: 10.3389/fmicb.2018.02147]
- Cai J, Sun L, Gonzalez FJ. Gut microbiota-derived bile acids in intestinal immunity, inflammation, and tumorigenesis. Cell Host Microbe 2022;30(3):289-300. [PMID: 35271802 DOI: 10.1016/j.chom.2022.02.004]
- Parida S, Sharma D. The power of small changes: comprehensive analyses of microbial dysbiosis in breast cancer. Biochim Biophys Acta Rev Cancer 2019;1871(2):392-405. [PMID: 30981803 DOI: 10.1016/j.bbcan.2019.04.001]
- Aarnoutse R, Ziemons J, Penders J, Rensen SS, de Vos-Geelen J, et al. The clinical link between human intestinal microbiota and systemic cancer therapy. Int J Mol Sci 2019;20(17):4145. [PMID: 31450659 DOI: 10.3390/ijms20174145]
- Saad R, Rizkallah MR, Aziz RK. Gut pharmacomicrobiomics: the tip of an iceberg of complex interactions between drugs and gut-associated microbes. Gut Pathog 2012;4(1):16. [PMID: 23194438 DOI: 10.1186/1757-4749-4-16]
- Montassier E, Batard E, Massart S, Gastinne T, Carton T, et al. 16S rRNA gene pyrosequencing reveals shift in patient faecal microbiota during high-dose chemotherapy as conditioning regimen for bone marrow transplantation. Microb Ecol 2014;67(3):690-9. [PMID: 24402367 DOI: 10.1007/s00248-013-0355-4]
- Zwielehner J, Lassl C, Hippe B, Pointner A, Switzeny OJ, et al. Changes in human fecal microbiota due to chemotherapy analyzed by TaqMan-PCR, 454 sequencing and PCR-DGGE fingerprinting. PLoS One 2011;6(12):e28654. [PMID: 22194876 DOI: 10.1371/journal.pone.0028654]
- Munyaka PM, Eissa N, Bernstein CN, Khafipour E, Ghia JE. Antepartum antibiotic treatment increases offspring susceptibility to experimental colitis: a role of the gut microbiota. PLoS One 2015;10(11):e0142536. [PMID: 26605545 DOI: 10.1371/journal.pone.0142536]
- Edgar RC. UPARSE: highly accurate OTU sequences from microbial amplicon reads. Nat Methods 2013;10(10):996-8. [PMID: 23955772 DOI: 10.1038/nmeth.2604]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol 2009;75(23):7537-41. [PMID: 19801464 DOI: 10.1128/AEM.01541-09]
- An J, Kwon H, Kim YJ. The firmicutes/bacteroidetes ratio as a risk factor of breast cancer. J Clin Med 2023;12(6):2216. [PMID: 36983217 DOI: 10.3390/jcm12062216]
- Bashiardes S, Tuganbaev T, Federici S, Elinav E. The microbiome in anti-cancer therapy. Semin Immunol 2017;32:74-81. [PMID: 28431920 DOI: 10.1016/j.smim.2017.04.001]
- Burcelin R, Serino M, Chabo C, Garidou L, Pomié C, et al. Metagenome and metabolism: the tissue microbiota hypothesis. Diabetes Obes Metab 2013;15(Suppl 3): 61-70. [PMID: 24003922 DOI: 10.1111/dom.12157]
- Jaye K, Li CG, Chang D, Bhuyan DJ. The role of key gut microbial metabolites in the development and treatment of cancer. Gut Microbes 2022;14(1):2038865. [PMID: 35220885 DOI: 10.1080/19490976.2022.2038865]
- Kovács T, Mikó E, Vida A, Sebo E, Toth J, et al. Cadaverine, a metabolite of the microbiome, reduces breast cancer aggressiveness through trace amino acid receptors. Sci Rep 2019;9(1):1300. [DOI: 10.1038/s41598-018-37664-7]
- Cao ZG, Qin XB, Liu FF, Zhou LL. Tryptophan-induced pathogenesis of breast cancer. Afr Health Sci 2015;15(3):982-5. [PMID: 26957990 DOI: 10.4314/ahs.v15i3.36]
- Bekki K, Vogel H, Li W, Ito T, Sweeney C, et al. The aryl hydrocarbon receptor (AhR) mediates resistance to apoptosis induced in breast cancer cells. Pestic Biochem Physiol 2015;120:5-13. [PMID: 25987214 DOI: 10.1016/j.pestbp.2014.12.021]
- Postler TS, Ghosh S. Understanding the holobiont: how microbial metabolites affect human health and shape the immune system. Cell Metab 2017;26(1):110-30. [PMID: 28625867 DOI: 10.1016/j.cmet.2017.05.008]
- Luu TH, Michel C, Bard JM, Dravet F, Nazih H, et al. Intestinal proportion of Blautia sp. is associated with clinical stage and histoprognostic grade in patients with early-stage breast cancer. Nutr Cancer 2017;69(2):267-75. [PMID: 28094541 DOI: 10.1080/01635581.2017.1263750]
- Fernández MF, Reina-Pérez I, Astorga JM, Rodríguez-Carrillo A, Plaza-Díaz J, et al. Breast cancer and its relationship with the microbiota. Int J Environ Res Public Health 2018;15(8):1747. [PMID: 30110974 DOI: 10.3390/ijerph15081747]
- Mikó E, Kovács T, Sebo É, Tóth J, Csonka T, et al. Microbiome-microbial metabolome-cancer cell interactions in breast cancer-familiar, but unexplored. Cells 2019;8(4):293. [PMID: 30934972 DOI: 10.3390/cells8040293]
- Alon-Maimon T, Mandelboim O, Bachrach G. Fusobacterium nucleatum and cancer. Periodontol 2000 2022;89(1):166-80. [PMID: 35244982 DOI: 10.1111/prd.12426]
- Duggan WP, Salvucci M, Kisakol B, Lindner AU, Reynolds IS, et al. Increased Fusobacterium tumoural abundance affects immunogenicity in mucinous colorectal cancer and may be associated with improved clinical outcome. J Mol Med (Berl) 2023;101(7):829-41. [PMID: 37171483 DOI: 10.1007/s00109-023-02324-5]
- Guo X, Yu K, Huang R. The ways Fusobacterium nucleatum translocate to breast tissue and contribute to breast cancer development. Mol Oral Microbiol 2024;39(1):1-11. [PMID: 38171827 DOI: 10.1111/omi.12446]
- Parhi L, Alon-Maimon T, Sol A, Nejman D, Shhadeh A, et al. Breast cancer colonization by Fusobacterium nucleatum accelerates tumor growth and metastatic progression. Nat Commun 2020;11(1):3259. [DOI: 10.1038/s41467-020-16967-2]
- Van der Merwe M, Van Niekerk G, Botha A, Engelbrecht AM. The onco-immunological implications of Fusobacterium nucleatum in breast cancer. Immunol Lett 2021;232:60-6. [PMID: 33647328 DOI: 10.1016/j.imlet.2021.02.007]
- Li G, Sun Y, Huang Y, Lian J, Wu S, et al. Fusobacterium nucleatum-derived small extracellular vesicles facilitate tumor growth and metastasis via TLR4 in breast cancer. BMC Cancer 2023;23(1):473. [PMID: 37221488 DOI: 10.1186/s12885-023-10844-z]
- Wang X, Fang Y, Liang W, Wong CC, Qin H, et al. Fusobacterium nucleatum facilitates anti-PD-1 therapy in microsatellite stable colorectal cancer. Cancer Cell 2024;42(10):1729-46.e8. [PMID: 39303724 DOI: 10.1016/j.ccell.2024.08.019]
- Gao Y, Bi D, Xie R, Li M, Guo J, et al. Fusobacterium nucleatum enhances the efficacy of PD-L1 blockade in colorectal cancer. Signal Transduct Target Ther 2021;6(1):398. [PMID: 34795206 DOI: 10.1038/s41392-021-00795-x]
- Chen Z, Huang L. Fusobacterium nucleatum carcinogenesis and drug delivery interventions. Adv Drug Deliv Rev 2024;209:115319. [PMID: 38643839 DOI: 10.1016/j.addr.2024.115319]
- Liu Y, Baba Y, Ishimoto T, Tsutsuki H, Zhang T, et al. Fusobacterium nucleatum confers chemoresistance by modulating autophagy in oesophageal squamous cell carcinoma. Br J Cancer 2021;124(5):963-74. [PMID: 33299132 DOI: 10.1038/s41416-020-01198-5]
- Zeng W, Pan J, Ye G. miR-135b aggravates fusobacterium nucleatum-induced cisplatin resistance in colorectal cancer by targeting KLF13. J Microbiol 2024;62(2):63-73. [PMID: 38402337 DOI: 10.1007/s12275-023-00100-1]
- Lu ZN, Song J, Song J, Sun TH, Sun G. UBE2C affects breast cancer proliferation through the AKT/mTOR signaling pathway. Chin Med J (Engl) 2021;134(20):2465-74. [PMID: 34620747 DOI: 10.1097/CM9.0000000000001708]
- Miricescu D, Totan A, Stanescu-Spinu II, Badoiu SC, Stefani C, et al. PI3K/AKT/mTOR signaling pathway in breast cancer: from molecular landscape to clinical aspects. Int J Mol Sci 2020;22(1):173. [PMID: 33375317 DOI: 10.3390/ijms22010173]
- Hanker AB, Sudhan DR, Arteaga CL. Overcoming endocrine resistance in breast cancer. Cancer Cell 2020;37(4):496-513. [PMID: 32289273 DOI: 10.1016/j.ccell.2020.03.009]
- Saatci O, Huynh-Dam KT, Sahin O. Endocrine resistance in breast cancer: from molecular mechanisms to therapeutic strategies. J Mol Med (Berl) 2021;99(12):1691-710. [PMID: 34623477 DOI: 10.1007/s00109-021-02136-5]
- Kulkoyluoglu-Cotul E, Arca A, Madak-Erdogan Z. Crosstalk between estrogen signaling and breast cancer metabolism. Trends Endocrinol Metab 2019;30(1):25-38. [PMID: 30471920 DOI: 10.1016/j.tem.2018.10.006]
- Saha T, Makar S, Swetha R, Gutti G, Singh SK. Estrogen signaling: an emanating therapeutic target for breast cancer treatment. Eur J Med Chem 2019;177:116-43. [PMID: 31129450 DOI: 10.1016/j.ejmech.2019.05.023]
- Zhang J, Zhang M, Huo XK, Ning J, Yu ZL, et al. Macrophage inactivation by small molecule wedelolactone via targeting sEH for the treatment of LPS-induced acute lung injury. ACS Cent Sci 2023;9(3):440-56. [PMID: 36968547 DOI: 10.1021/acscentsci.2c01424]
- Sun CP, Zhou JJ, Yu ZL, Huo XK, Zhang J, et al. Kurarinone alleviated Parkinson’s disease via stabilization of epoxyeicosatrienoic acids in animal model. Proc Natl Acad Sci U S A 2022;119(9):e2118818119. [PMID: 35217618 DOI: 10.1073/pnas.2118818119]
- Zhang J, Luan ZL, Huo XK, Zhang M, Morisseau C, et al. Direct targeting of sEH with alisol B alleviated the apoptosis, inflammation, and oxidative stress in cisplatin-induced acute kidney injury. Int J Biol Sci 2023;19(1):294-310. [PMID: 36594097 DOI: 10.7150/ijbs.78097]
- Zhang J, Zhang WH, Morisseau C, Zhang M, Dong HJ, et al. Genetic deletion or pharmacological inhibition of soluble epoxide hydrolase attenuated particulate matter 2.5 exposure mediated lung injury. J Hazard Mater 2023;458:131890. [PMID: 37406527 DOI: 10.1016/j.jhazmat.2023.131890]
- Jia YX, Wang N, Hui SW, Chang J, Zhu QM, et al. Discovery of soluble epoxide hydrolase inhibitors from Inula britannica: inhibition kinetics, molecular dynamics simulation, biochemical, and in vitro cell-based studies. Int J Biol Macromol 2025;306(Pt 3):141704. [DOI: 10.1016/j.ijbiomac.2025.141704]
- Zhang HL, Wang N, Shi XL, Wang MM, Zhu QM, et al. Sesquiterpenoids from Inula britannica and their potential mechanism for immunomodulation. Phytochemistry 2025;231:114343. [PMID: 39613278 DOI: 10.1016/j.phytochem.2024.114343]