The metabolite 2-Methylbutyrylcarnitine does not Promote Atherosclerosis in Apolipoprotein E-Deficient Mice
1Guangdong Provincial Key Laboratory of Malignant Tumor Epigenetics and Gene Regulation, Guangdong-Hong Kong Joint Laboratory for RNA Medicine, Medical Research Center, Sun Yat-Sen Memorial Hospital, Sun Yat-Sen University, Guangzhou, Guangdong 510120, China
2Nanhai Translational Innovation Center of Precision Immunology, Sun Yat-Sen Memorial Hospital, Foshan, Guangdong 528200, China
3Division of Vascular Surgery, The First Affiliated Hospital of Sun Yat-Sen University, Guangzhou, Guangdong 510080, China
4Department of Endocrinology, Sun Yat-Sen Memorial Hospital, Guangzhou, Guangdong 510120, China
aThese authors contributed equally.
*Correspondence to: Sifan Chen, E-mail: chensf26@mail.sysu.edu.cn
Received: July 7 2024; Revised: July 21 2024; Accepted: July 23 2024; Published Online: July 30 2024
Cite this paper:
Wen J, Zhao Z, Cen Z et al. The metabolite 2-Methylbutyrylcarnitine does not Promote Atherosclerosis in Apolipoprotein E-Deficient Mice. BIO Integration 2024; 5: 1–9.
DOI: 10.15212/bioi-2024-0049. Available at: https://bio-integration.org/
Download citation
© 2024 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Background: Although 2-methylbutyrylcarnitine (2MBC) has been associated with metabolic disorders and promotes thrombosis, its effect on atherosclerosis remains elusive. This study was aimed at investigating the role of 2MBC in atherosclerosis development.
Methods and Results: Apolipoprotein E-deficient (ApoE−/−) mice were fed a Western diet for 18 weeks to induce atherosclerosis, then administered once-daily gavage with 2MBC or vehicle for 18 weeks. Parameters of systemic lipid metabolism and atherosclerosis were detected. Although 2MBC did not upregulate plasma total cholesterol (TC), low-density lipoprotein cholesterol (LDL-C), and high-density lipoprotein cholesterol (HDL-C) levels, the plasma total triglycerides (TG) levels were further upregulated in Western-diet-induced mice treated with 2MBC, thus suggesting that this compound may contribute to hypertriglyceridemia. In addition, 2MBC had no effect on atherosclerosis development, as evidenced by no alterations in plaque area, exacerbation of local inflammation, or effects on plaque stability. RAW264.7 macrophages were used to investigate the effect of 2MBC on oxidized low-density lipoprotein-induced foam cell formation in vitro. Treatment with 2MBC did not affect lipid uptake by foam cells. The addition of 2MBC did not affect the relative mRNA levels of inflammation-associated genes when macrophages were treated with lipopolysaccharide. In addition, to investigate the destructive effects of 2MBC on the vascular endothelium, we stimulated human umbilical vein endothelial cells (HUVECs) with oxidized low-density lipoprotein (ox-LDL). Ox-LDL did not alter the expression levels of monocyte chemotactic protein-1 or vascular cell adhesion molecule-1. Furthermore, 2MBC combined with ox-LDL stimulation did not alter the expression of SR-A1 and ABCA1 in HUVECs.
Conclusions: Our study provides the first evidence that 2MBC does not promote atherosclerosis development. This compound does not increase intravascular plaque area, exacerbate the degree of local inflammation, or affect plaque stability in ApoE−/− mice.
Keywords
2-Methylbutyrylcarnitine, atherosclerosis, atherosclerotic plaques.
Statement of Significance
The causal relationship between 2MBC and atherosclerosis remains unclear. Our findings demonstrate that 2MBC does not promote disease progression in atherosclerosis.
Introduction
Atherosclerosis is responsible for most cardiovascular diseases and is a leading cause of adult morbidity and mortality worldwide [1, 2]. Atherosclerosis is considered a result of metabolic disorders, and is characterized by the focal retention and accumulation of fatty materials within arterial walls in the form of plaques [3, 4]. The progression of atherosclerosis ultimately leads to narrowing and hardening of the arterial lumen with impaired and restricted blood flow [5, 6]. Subsequent plaque rupture and atherosclerotic thrombotic occlusion of vessels greatly predispose individuals to life-threatening cardiovascular events, notably ischemic stroke and myocardial infarction. Consequently, exploring the mechanism underlying atherosclerosis is particularly important.
Branched chain amino acids (BCAAs), essential amino acids required by mammals, are obtained primarily from dietary consumption and biosynthesis by gut microorganisms [7]. Numerous findings suggest a correlation between the dysregulation of circulating BCAAs and their metabolites, and disorders of glycolipid metabolism in the body [8–11]. Moreover, a link has been demonstrated between elevated BCAA levels and cardiovascular disease states; elevated plasma BCAA concentrations are biomarkers of heart failure, coronary artery disease, and hypertension, and can even predict poor patient outcomes [12]. With increasing BCAA intake, plaque volume, plaque instability, and inflammation are significantly elevated in ApoE−/− mice [13]. As a member of the short-branched-chain acylcarnitine family, 2MBC is an intermediate product of BCAA catabolism [14–17]. Several studies have revealed associations between short-branched-chain acylcarnitine and various metabolic disorders, including diabetes, obesity, and non-alcoholic steatohepatitis [18–21]. The results of our previous study have indicated that upregulated plasma 2MBC levels in patients with thrombosis are positively associated with thrombotic risk in humans. The compound 2MBC binds integrin α2β1 in platelets, thereby potentiating cytosolic phospholipase A2 (cPLA2) activation and platelet hyperresponsiveness [22].
Atherosclerotic plaques lead to significant clinical events when they limit myocardial blood flow via progressive stenosis or thrombus formation. The formation and thickening of plaques, which predispose individuals to the inexorable outcome of thrombosis, has prompted questions regarding whether 2MBC, as a pro-thrombotic factor, might also play crucial roles in the development and progression of atherosclerosis. In line with this hypothesis, the potential role and associated mechanism of 2MBC in atherosclerosis were investigated in this study.
Methods
Animal model
All animal experiments were performed in accordance with the Guidelines for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committees of Sun Yat-Sen Memorial Hospital (approval number AP20230069). To avoid potential confounding effects associated with female sex, our in vivo experiments used 6-week-old male apolipoprotein E-deficient (ApoE−/−) mice on a C57BL/6J background and their littermate wild-type controls, obtained from GemPharmatech Co. Ltd (Nanjing, China). All mice were housed under specific-pathogen-free conditions in a controlled environment (23±2°C with a relative humidity of 50%–60% and a 12-hour day/night cycle), and were given free access to food and water. After 1 week of acclimation, the ApoE−/− mice were randomly divided into the following four groups, with eight mice each: normal diet (ND) group, ND+2MBC group, Western diet (WD) group, and WD+2MBC group. The WD and WD+2MBC groups were fed a diet containing 45% fat (kcal/100 g) and 0.2% (wt/wt) cholesterol (TP26300, TROPHIC, Nantong, China) for 18 weeks. The ND and ND+2MBC groups were fed a low-sugar, low-fat control diet (TP26312, TROPHIC, Nantong, China) for 18 weeks. During the feeding period, the mice were administered 0.39 mg/kg 2MBC (Sigma-Aldrich, St. Louis, MO, USA) or H2O daily by gavage. The wild-type control mice (n=5) were simultaneously fed the control diet for 18 weeks. Food intake was recorded daily, and body weight was monitored weekly. After a 6-hour fast at the end of the study, the mice were anesthetized with intraperitoneal sodium pentobarbital (80 mg/kg), and blood was collected from the retro-orbital plexus. Tissue samples were preserved in 4% paraformaldehyde for subsequent analysis.
Lipid profile analysis
Levels of total cholesterol, total triglycerides, low-density lipoprotein cholesterol, and high-density lipoprotein cholesterol levels in the plasma were determined with commercial assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing, China).
Histology and analysis
Atherosclerotic lesions were evaluated according to established methods [23]. Briefly, ice-cold PBS was perfused into the vasculature via apical left ventricular puncture. Subsequently, the aorta and heart were separated and removed immediately. To assess plaque size at the aortic root, we cut samples in the ascending aorta, and embedded the proximal samples containing the aortic roots in optimal cutting temperature compound. Serial sections (7 μm thickness) were collected from each mouse and processed for hematoxylin and eosin, Oil Red O, and Masson trichrome staining according to standardized protocols. The areas of plaques, Oil Red O-positivity, and collagen were measured in ImageJ software. In detail, plaque size was determined as the area between the lumen border and the internal elastic lamina border in the aortic root. Digital images of stained sections were captured, and the percentage stained area (stained area per total atherosclerotic lesion area) was determined.
Cell culture and experimental conditions
HUVECs were obtained from the National Collection of Authenticated Cell Cultures (Shanghai, China) and cultured in F-12K medium (Hyclone, South Logan, UT, USA) containing 10% fetal bovine serum (Gibco, Beijing, China), heparin (0.1 mg/mL), and Endothelial Cell Growth Supplement (30 μg/mL). The medium also contained penicillin and streptomycin (100 IU/mL each). RAW264.7 cells were acquired from the National Collection of Authenticated Cell Cultures and maintained in RPMI-1640 medium (Gibco, Beijing, China) supplemented with 10% fetal bovine serum (Gibco, Paisley, Scotland, UK). All cell lines were cultured at 37°C in a 5% CO2 cell culture incubator, and cells were used until passage 15. RAW264.7 or HUVEC cells were co-incubated with ox-LDL (100 μg/mL; Yiyuan Biotech, Guangzhou, China) or lipopolysaccharide (100 ng/ml; Yiyuan Biotech, Guangzhou, China) for 24 hours, with simultaneous addition of 2MBC (final concentration 0.5 μM) or H2O.
Oil red O staining
RAW264.7 cells were fixed with 4% paraformaldehyde for 30 minutes. After fixation, the cells were rinsed twice with PBS, then stained with filtered Oil Red O solution (Sigma-Aldrich, St. Louis, MO, USA) at room temperature for 15 minutes. Subsequently, cells were destained with 60% isopropyl alcohol for 5 seconds. Oil Red O staining was visualized under a light microscope, and the intensity was measured in ImageJ software (National Institutes of Health, Bethesda, MD).
RNA extraction and quantitative real-time reverse transcription polymerase chain reaction
Total RNA was isolated with RNAex Pro Reagent (Accurate Biology, Hunan, China), and 1 μg total RNA was reverse transcribed to a cDNA template with an Evo M-MLVRT kit (Accurate Biology, Hunan, China). The primer sequences were deduced from PrimerBank and are listed in Table 1. Quantitative polymerase chain reaction (PCR) was performed with 1 μL cDNA obtained as described above in 10 μL volumes containing 5 mM primers, with a SYBR Green Premix Pro Taq HS qPCR Kit (Accurate Biology, Hunan, China) on a LightCycler 480 system. The PCR conditions were as follows: 95°C for 5 minutes, 45 cycles at 95°C for 10 seconds, and 60°C for 10 seconds. PCR results were normalized to the expression of GAPDH in the same samples.
Table 1 Polymerase Chain Reaction Primers Used for Amplification
| Species | Gene | Primers |
|---|---|---|
| Human | ICAM-1 | 5′-AGCGGCTGACGTGTGCAGTAAT-3′ |
| 5′-TCTGAGACCTCTGGCTTCGTCA-3′ | ||
| MCP-1 | 5′-AGAATCACCAGCAGCAAGTGTCC-3′ | |
| 5′-TCCTGAACCCACTTCTGCTTGG-3′ | ||
| Interleukin-1β | 5′-CCACAGACCTTCCAGGAGAATG-3′ | |
| 5′-GTGCAGTTCAGTGATCGTACAGG-3′ | ||
| TNF-α | 5′-CTCTTCTGCCTGCTGCACTTTG-3′ | |
| 5′-ATGGGCTACAGGCTTGTCACTC-3′ | ||
| Interleukin-6 | 5′-AGACAGCCACTCACCTCTTCAG-3′ | |
| 5′-TTCTGCCAGTGCCTCTTTGCTG-3′ | ||
| Interleukin-8 | 5′-GAGAGTGATTGAGAGTGGACCAC-3′ | |
| 5′-CACAACCCTCTGCACCCAGTTT-3′ | ||
| SR-A1 | 5′-GCAGTGGGATCACTTTCACAA-3′ | |
| 5′-AGCTGTCATTGAGCG AGCATC-3′ | ||
| ABCA1 | 5′-CAGGCTACTACCTGACCTTGGT-3′ | |
| 5′-CTGCTCTGAGAAACACTGTCCTC-3′ | ||
| Mouse | Cd36 | 5′-GGACATTGAGATTCTTTTCCTCTG-3′ |
| 5′-GCAAAGGCATTGGCTGGAAGAAC-3′ | ||
| SR-A1 | 5′-CGCACGTTCAATGACAGCATCC-3′ | |
| 5′-GCAAACACAAGGAGGTAGAGAGC-3′ | ||
| LOX-1 | 5′-GTCATCCTCTGCCTGGTGTTGT-3′ | |
| 5′-TGCCTTCTGCTGGGCTA ACATC-3′ | ||
| Interleukin-1β | 5′-TGGACCTTCCAGGATGAGGACA-3′ | |
| 5′-GTTCATCTCGG AGCCTGTAGTG-3′ | ||
| TNF-α | 5′-GGTGCCTATGTCTCAGCCTCTT-3′ | |
| 5′-GCCATAGAACTGATGAGAGGGAG-3′ | ||
| MCP-1 | 5′-CCACTCACCTGCTGCTACTCA-3′ | |
| 5′-TGGTGATCCTCTTGTAGCTCTCC-3′ |
Abbreviations: ICAM-1, intercellular cell adhesion molecule-1; MCP-1, monocyte chemoattractant protein-1; TNF-α, tumor necrosis factor-α; SR-A1, scavenger receptor A1; ABCA1, ATP-binding cassette transporter A1; and LOX-1, lectin-like oxidized low-density lipoprotein receptor-1.
Statistical analysis
All statistical analyses were conducted in Prism 8 (GraphPad Software, La Jolla, CA), and numerical values are presented as mean±SEM. Representative data are displayed. For comparisons between two groups, the homogeneity of variance was first assessed, and the significance of differences was determined with Student’s t-test. For samples with three groups or more, one-way analysis of variance followed by Tukey’s multiple comparison test was used for statistical analysis. Significance levels are typically represented as follows: ns, no significant difference; *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001. The letter “n” in the figures denotes the number of experimental repetitions or the sample size, and corresponding p-values and “n” values are provided in the respective figure legends.
Results
2MBC plays an insignificant role in promoting lipid disorders
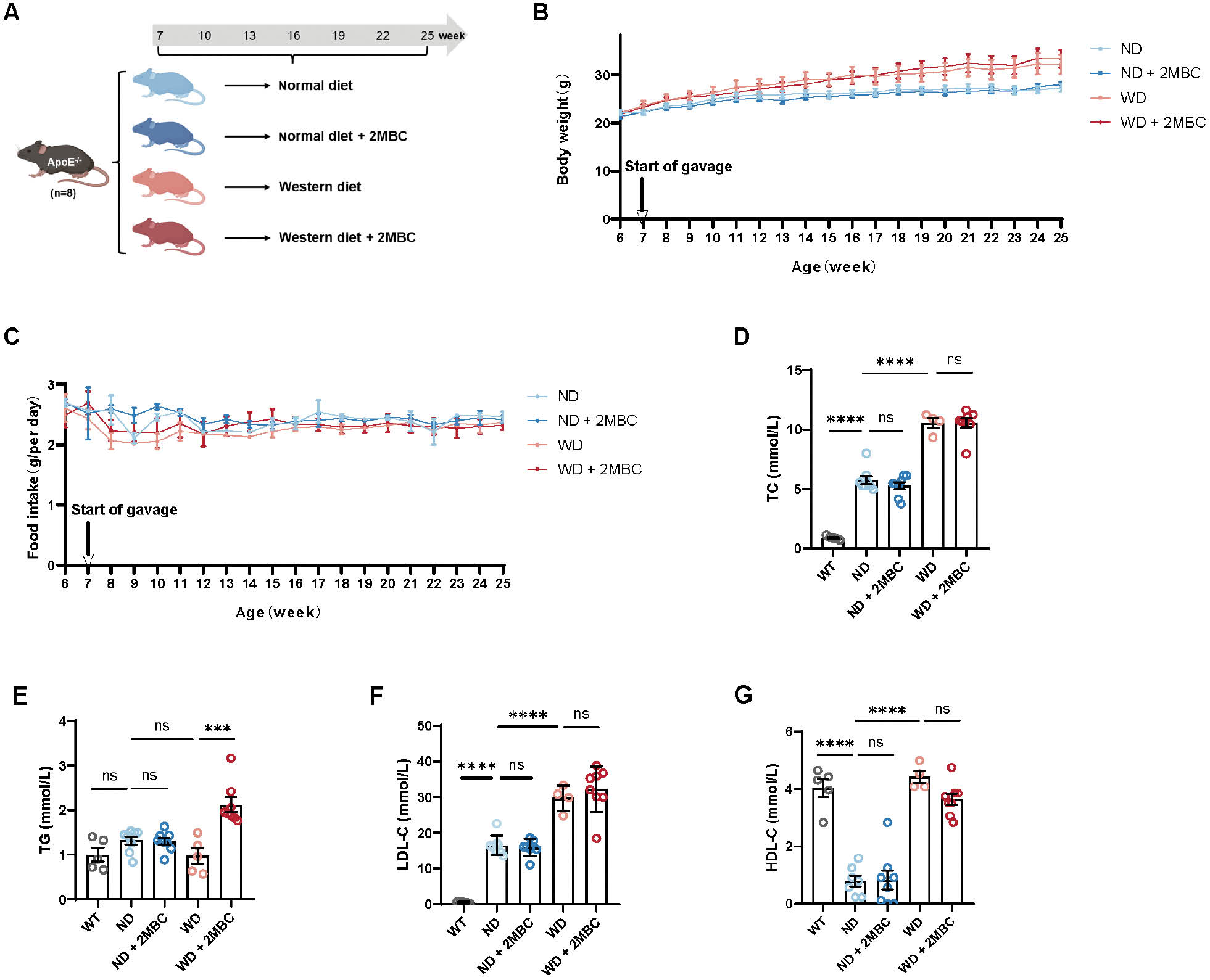
Atherosclerosis animal models are an indispensable tool for studying atherosclerotic plaque formation and progression; among these models, ApoE−/− mice are widely used [24, 25]. The glycoprotein ApoE functions as a ligand for receptors that scavenge chyle granules and VLDL residues. Deficiency in this glycoprotein can lead to increases in total plasma cholesterol; therefore, ApoE is a powerful tool to study lipid metabolic diseases and atherosclerosis. To further investigate the effects of 2MBC treatment on systemic metabolism, we fed male ApoE−/− mice a WD or ND, and administered 2MBC or vehicle by gavage for 14 weeks (Figure 1A). Administration of 2MBC did not alter in body weight (Figure 1B) or daily food intake among the four groups (Figure 1C). In addition, plasma TC and LDL-C levels were markedly higher in ND-fed ApoE−/− mice than wild-type (WT) mice, and were further elevated in WD-fed ApoE−/− mice (Figure 1D, F). In contrast, plasma HDL-C was significantly lower in ND-fed ApoE−/− mice than WT mice, whereas plasma HDL-C was elevated in WD-fed ApoE−/− mice (Figure 1G). However, contrary to our expectations, plasma lipid levels were not affected by 2MBC administration. The plasma levels of TC, LDL-C, and HDL-C were not significantly altered in 2MBC gavage-treated ApoE−/− mice. Furthermore, plasma TG levels were not significantly elevated in ApoE−/− mice compared with WT mice (Figure 1E). Notably, after WD feeding and 2MBC gavage, the ApoE−/− mice exhibited substantially increased plasma TG levels. Together, our data illustrated that 2MBC does not promote dysfunctional lipid metabolism.
Figure 1 2MBC plays an insignificant role in promoting lipid disorders. After 18 weeks of Western diet feeding, ApoE−/− mice were randomly divided into four groups and subsequently treated with 2MBC or vehicle for 18 weeks. A, Animal experimental design; B, body weight changes; and C, daily food intake after intervention. n=8 for each group. D, Plasma TC; E, plasma TG; F, plasma LDL-C; G, plasma HDL-C. Error bars represent SEM. ***p<0.001; ****p<0.0001 vs. control. TC, total cholesterol; TG, total triglycerides; LDL-C, low-density lipoprotein cholesterol; HDL-C, high-density lipoprotein cholesterol..
2MBC does not promote lipid accumulation within plaques
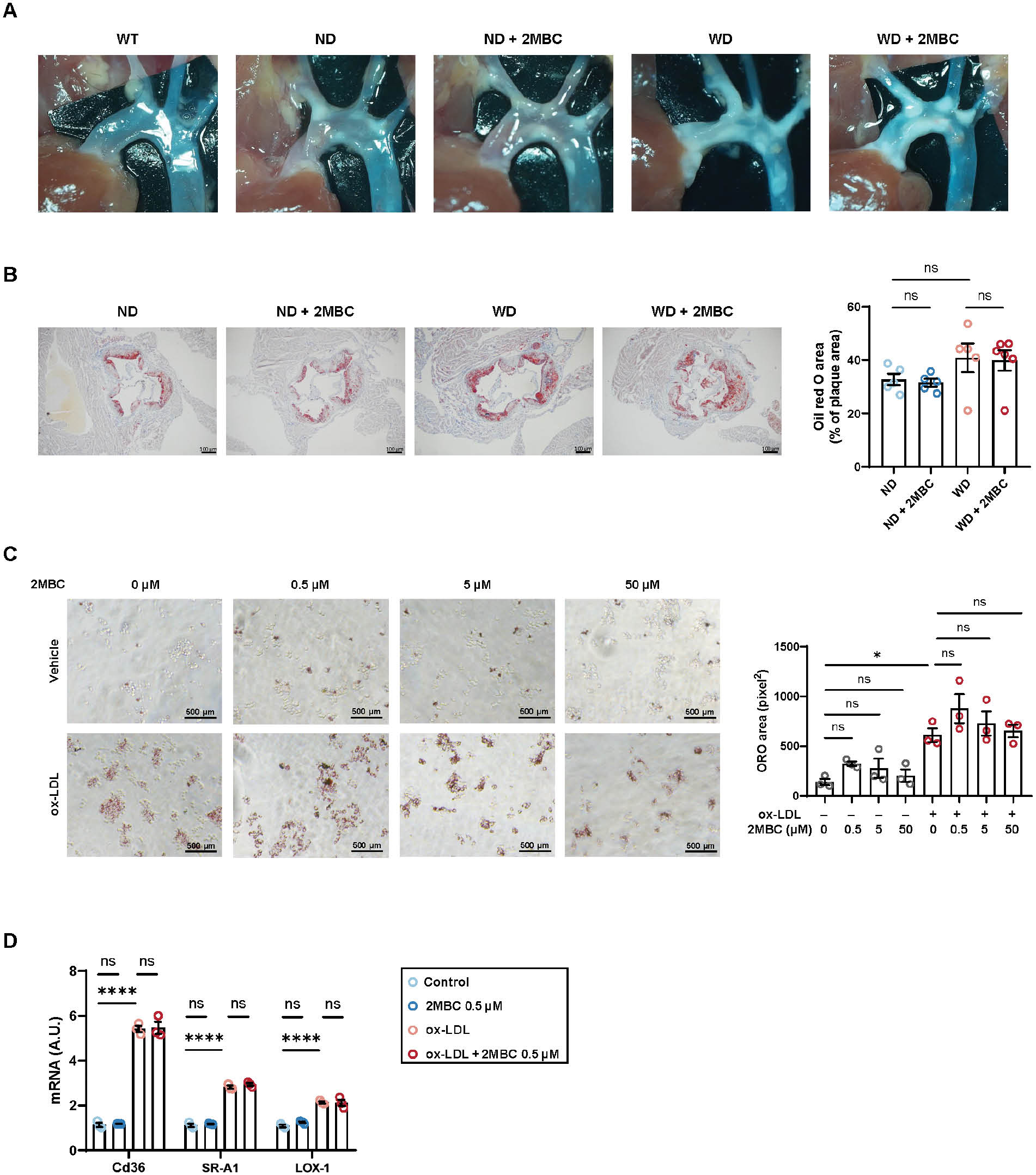
Lipid accumulation trend was observed in the aortic root of ApoE–/– mice compared with WT mice, and even greater lipid deposition was observed in WD mice; however, we were not able to directly visualize the role of 2MBC on atheromatous plaque formation (Figure 2A). To determine whether 2MBC might exacerbate atherosclerosis injury in vivo, and to assess aortic root plaque lesions, we performed Oil Red O staining of aortic root sections to assess the extent of intravascular lipid deposition. The lipid area was 8.1% greater in the WD group than the normal diet group (Figure 2B). However, the increase in 2MBC did not increase intravascular lipid area in either dietary setting. Many studies have shown that high plasma LDL levels are a prominent risk factor for atherosclerosis. In atherosclerosis progression, LDL tends to accumulate in the subendothelial space of the arterial wall and to undergo gradual oxidative modification, thereby forming oxidized LDL (ox-LDL). Given the important role of macrophage-derived foam cells in atherosclerosis development in vivo, we explored whether 2MBC might play a role in ox-LDL-induced macrophage-derived foam cell formation through in vitro experiments. Oil Red O staining revealed that ox-LDL-induced foam cells contained more lipids than uninduced cells, but the 2MBC intervention did not increase the intracellular lipid content (Figure 2C). In addition, we analyzed genes involved in macrophage uptake of oxidized-LDL particles, including Cd36, SR-A1, and LOX-1, none which showed altered expression (Figure 2D). Collectively, our results suggested that 2MBC does not aggravate intravascular lipid deposition.
Figure 2 2MBC does not promote lipid accumulation within plaques. A, Representative photograph of aorta in vivo. B, Representative photographs of Oil Red O staining in the aortic root and quantitative analyses of Oil Red O area. RAW264.7 macrophages with or without 2MBC treatment were stimulated with ox-LDL for 24 h. Scale bars, 100 μm. C, Representative Oil Red O staining and quantitative analyses of Oil Red O. Scale bars, 500 μm. D, Relative mRNA expression of genes involved in ox-LDL uptake; GAPDH mRNA was used as an internal control. Data are representative of three independent experiments and are shown as mean±SEM. *p<0.05; ****p<0.0001 vs. control. SR-A1, scavenger receptor A1; LOX-1, lectin-like oxidized low-density lipoprotein receptor-1.
2MBC does not exacerbate inflammation at plaques
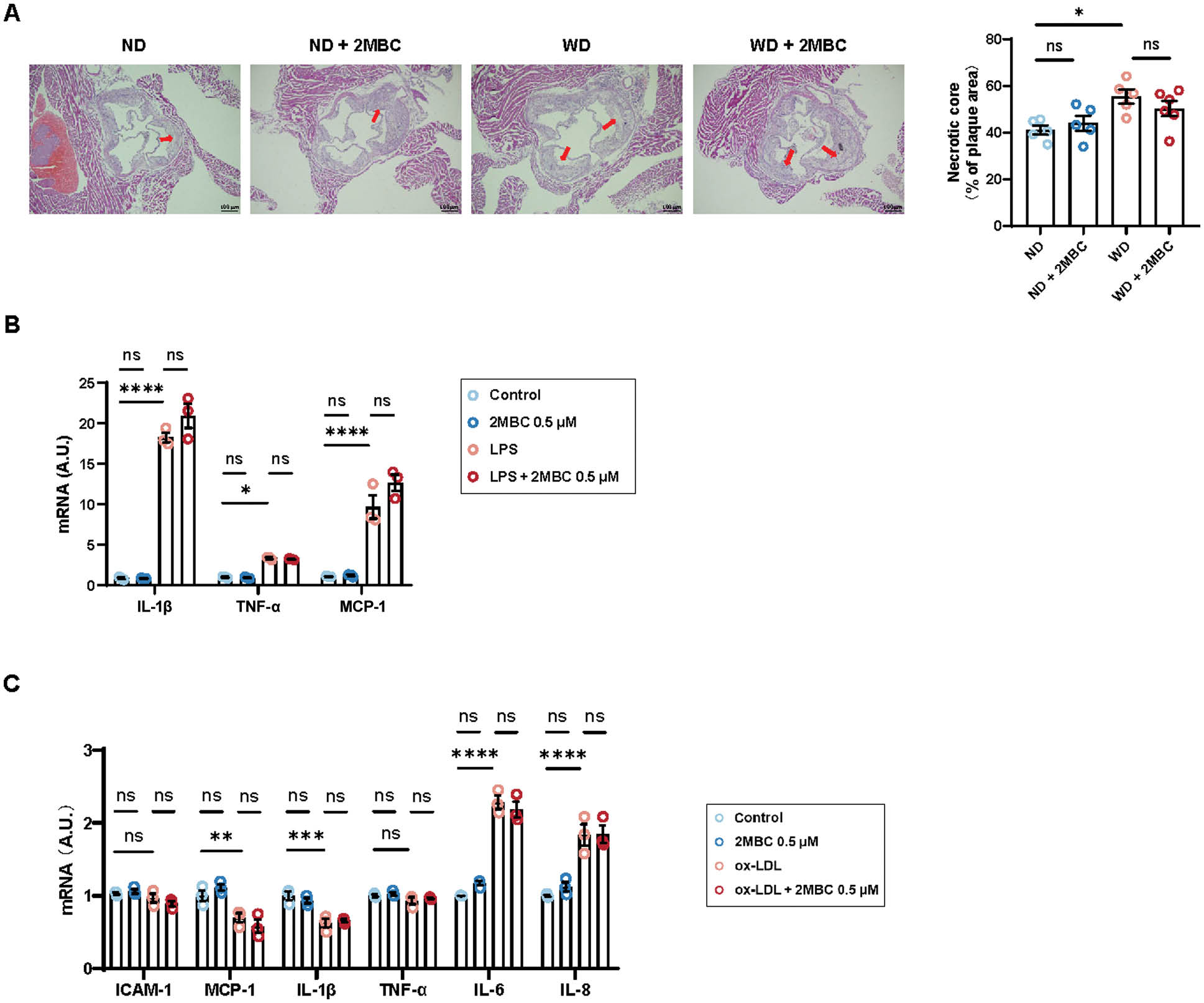
WD treatment resulted in a 14.5% increase in necrotic core area in ApoE−/− mice (Figure 3A). However, no significant differences in necrotic core size were observed between the 2MBC-treated group and the control group. We treated macrophages with LPS to mimic the inflammatory response environment in vivo and then additionally performed 2MBC treatment; however, no changes were observed in expression of IL-1β, TNF-α, and Mcp-1 (Figure 3B). In addition, ox-LDL induces endothelial cells to overexpress chemotactic proteins (such as monocyte chemotactic protein-1 (MCP-1)) and adhesion molecules (vascular cell adhesion molecule-1), thereby creating an inflammatory response environment. To investigate whether 2MBC might damage the vascular endothelium, we treated HUVECs with ox-LDL, with or without 2MBC. However, the expression of ICAM-1, MCP-1, IL-1β, TNF-α, IL-6, and IL-8 showed no significant changes, thus suggesting that 2MBC did not further promote inflammation in HUVECs (Figure 3C). Together, our data suggested that 2MBC does not promote inflammatory effects.
Figure 3 2MBC does not exacerbate inflammation at plaques. A, Representative photographs of H&E staining in the aortic root and quantitative analyses of necrotic core area. RAW264.7 macrophages with or without 2MBC treatment were stimulated with LPS for 24 h. Scale bars, 100 μm. B, Relative mRNA expression of genes involved in inflammation. HUVECs with or without 2MBC treatment were stimulated with ox-LDL for 24 h. C, Relative mRNA expression of genes involved in inflammation. GAPDH mRNA was used as an internal control. Data are representative of three independent experiments and are shown as mean±SEM. *p<0.05; **p<0.01; ***p<0.001; ****p<0.0001 vs control. IL-1β, interleukin-1β, TNF-α, tumor necrosis factor-α; MCP-1, monocyte chemoattractant protein 1; ICAM-1, intercellular cell adhesion molecule-1; IL-6, interleukin-6; and IL-8, interleukin-8.
2MBC does not increase plaque stability
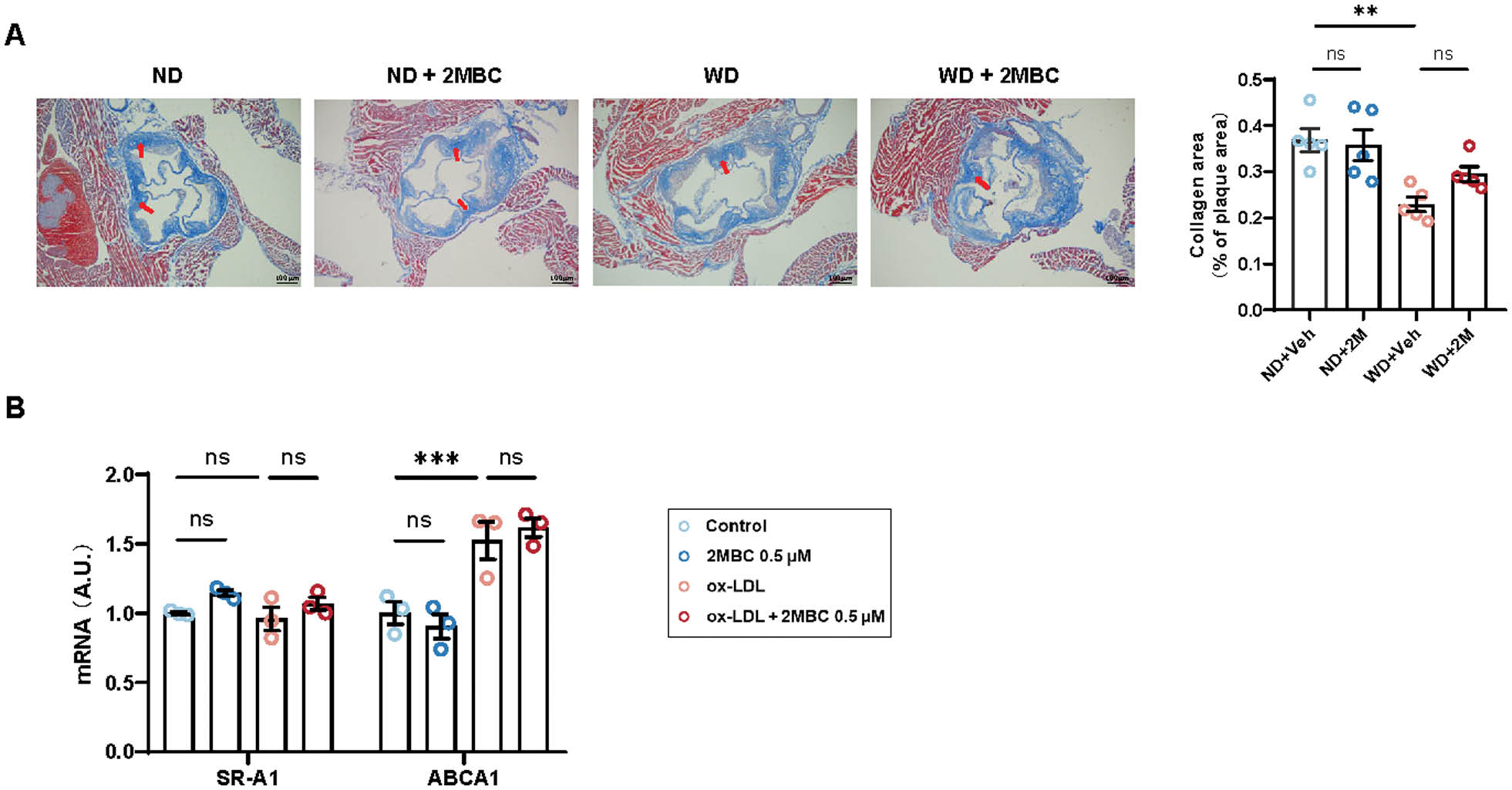
The formation of atherosclerotic plaques often leads to deceleration or even obstruction of blood flow. However, when an atherosclerotic plaque ruptures, the thrombogenic substances in the plaque are exposed, thus resulting in platelet aggregation, elevated plasma fibrinogen levels, and even myocardial infarction. Fragile plaques that break easily are characterized by large lipid cores and thin fibrous caps. A lower level of collagen in WD group, which was defined by Masson trichrome staining, indicated that the poor stability of the atherosclerotic plaque, compared with ND group (Figure 4A). However, we observed no effect on plaque stability after gavage with 2MBC. In addition, endothelial cells were treated with ox-LDL, and the mRNA levels of SR-A1 and ABCA1 remained unchanged in the presence or absence of 2MBC (Figure 4B). Therefore, we speculated that 2MBC does not affect plaque stability.
Figure 4 2MBC does not increase plaque stability. A, Representative photographs of Masson trichrome staining in the aortic root and quantitative analyses of collagen area. HUVECs with or without 2MBC treatment were stimulated with ox-LDL for 24 h. Scale bars, 100 μm. B, Relative mRNA expression of genes involved in cholesterol metabolism. GAPDH mRNA was used as an internal control. Data are representative of three independent experiments and are shown as mean±SEM. **p<0.01; ***p<0.001 vs control. ABCA1, ATP-binding cassette transporter A1.
Discussion
The regulatory role of 2MBC in the progression of atherosclerosis has not yet been investigated. Although several studies have reported that short-branched-chain acylcarnitines, represented by 2MBC, are upregulated in the context of metabolic disorders, the specific mechanisms and roles have not yet been studied. In the present study, although 2MBC did not upregulate plasma TC, LDL-C and HDL-C levels in mice, it significantly increased plasma TG levels in WD-fed mice. Numerous studies have shown an association between high TG values and adverse cardiovascular outcomes; thus, TG content testing is often used as an indicator of underlying atherosclerosis [26–28]. However, we found no significant effects of 2MBC on intravascular lipid accumulation, inflammatory response, and plaque stability. Both in vivo and in vitro results suggested that 2MBC might not contribute to the course and consequences of atherosclerosis development. In addition, plasma TG levels are strongly associated with the progression of several metabolic diseases, such as diabetes and fatty liver [29–31]. Although our results suggested that 2MBC-induced upregulation of plasma TG levels might not be associated with the progression of atherosclerosis, 2MBC might nonetheless play a role in the development and progression of certain metabolic diseases. This possibility warrants further investigation in further studies.
Atherosclerosis is a metabolic disorder involving atherogenic lipid deposition, pro-inflammatory conditions, and endothelial dysfunction [32]. Plasma LDL is oxidatively modified to ox-LDL, which in turn induces oxidative stress and inflammatory responses in vascular endothelial cells, and upregulates adhesion and chemotactic molecules such as intercellular adhesion molecule-1 and monocyte chemotactic protein-1 [33]. Moreover, foam cells secrete large amounts of inflammatory mediators in the vessel wall, thereby promoting arterial inflammation and driving smooth muscle cell migration from the mid-membrane to the subendothelial space, thus facilitating proliferation and differentiation [34]. The short-chain acylcarnitine 2MBC has a five-carbon chain, which is derived from the catabolism of BCAAs. Although current studies have shown no relationship between plasma 2MBC levels and atherosclerosis, elevated levels of 2MBC precursors such as BCAAs, are closely associated with the development and progression of atherosclerosis [12, 13]. However, 2MBC did not affect plasma LDL-C levels or foam cell formation, or promote inflammatory responses. These findings suggested that 2MBC might not have significant effects on systemic lipid metabolism or the pathophysiological processes of plaque formation and development, and is therefore unlikely to contribute to the progression of atherosclerosis.
As a pro-thrombotic factor, 2MBC is elevated in metabolic diseases, wherein platelets are chronically hyper-responsive. Up-regulation of 2MBC mobilizes more platelets to participate in the clotting process during thrombosis, thus increasing thrombosis severity [22]. Treatment with 2MBC showed no effect on vascular endothelial damage and intravascular lipid deposition—the primary characteristic of the pre-atherosclerotic stage of atherosclerosis. Therefore, we speculated that 2MBC does not contribute to atherosclerosis development. As the disease advances, plaques excessively accumulate in blood vessels, thereby blocking vessels and facilitating blood clot formation. The shedding of the plaque fibrous cap also activates blood clotting and results in thrombosis. Embolisms may be dislodged, travel with the blood flow, and consequently block other distant blood vessels [35]. At this stage, 2MBC may promote the rate of thrombosis and exacerbate thrombosis severity. Thus, in atherosclerosis, a prethrombotic disease, 2MBC presence may serve only as a marker of the magnitude of thrombotic risk, without having a causal regulatory role in atherosclerosis.
In conclusion, we report what is, to our knowledge the first investigation of the role of 2MBC in atherosclerosis development. Our studies demonstrated that 2MBC does not contribute atherosclerosis development by accelerating intravascular lipid accumulation, exacerbating inflammatory responses, or destabilizing intravascular plaques. However, 2MBC was found to upregulate plasma TG levels—an effect warranting further investigation in the future. Additionally, we acknowledge several limitations of our study: (1) the effects of 2MBC on atherosclerosis were not investigated in multiple animal models, and (2) the effects of various doses of 2MBC on atherosclerosis was not explored.
Author contributions
Conceptualization, supervision, project administration, funding acquisition, and writing—review and editing, S. C.; methodology, J. W., Z. Z., Z. C., and R. Z.; formal analysis, visualization, data curation, and writing—original draft preparation, J. W., Z. Z., L. L., L. Y., and Y. C.; formal analysis, and writing—original draft preparation, Z. C.
Funding
This work was supported by the National Natural Science Foundation of China (No. 32271185); National Natural Science Foundation of China Excellent Young Scientists Fund (No. 82222014); Guangzhou Science and Technology Program (2023A03J0706); Guangzhou Science and Technology Program Key Projects (No. 202206010031); Guangdong Basic and Applied Basic Research Foundation (No. 2021B1515020005); and Guangdong Science and Technology Department (2023B1212060013).
Conflicts of interest
The authors declare no conflicts of interest.
References
- Puylaert P, Zurek M, Rayner KJ, De Meyer GRY, Martinet W. Regulated necrosis in atherosclerosis. Arterioscler Thromb Vasc Biol 2022;42(11):1283-306. [DOI: 10.1161/atvbaha.122.318177]
- Jebari-Benslaiman S, Galicia-García U, Larrea-Sebal A, Olaetxea JR, Alloza I, et al. Pathophysiology of atherosclerosis. Int J Mol Sci 2022;23(6):3346. [PMID: 35328769 DOI: 10.3390/ijms23063346]
- Libby P, Buring JE, Badimon L, Hansson GK, Deanfield J, et al. Atherosclerosis. Nat Rev Dis Primer 2019;5(1):56. [PMID: 31420554 DOI: 10.1038/s41572-019-0106-z]
- Wu J, He S, Song Z, Chen S, Lin X, et al. Macrophage polarization states in atherosclerosis. Front Immunol 2023;14:1185587. [PMID: 37207214 DOI: 10.3389/fimmu.2023.1185587]
- Libby P. The changing landscape of atherosclerosis. Nature 2021;592(7855):524-33. [PMID: 33883728 DOI: 10.1038/s41586-021-03392-8]
- Nayor M, Brown KJ, Vasan RS. The molecular basis of predicting atherosclerotic cardiovascular disease risk. Circ Res 2021;128(2):287-303. [PMID: 33476202 DOI: 10.1161/CIRCRESAHA.120.315890]
- Li N, Cen Z, Zhao Z, Li Z, Chen S. BCAA dysmetabolism in the host and gut microbiome, a key player in the development of obesity and T2DM. Med Microecol 2023;16:100078. [DOI: 10.1016/j.medmic.2023.100078]
- Sánchez-González C, Nuevo-Tapioles C, Herrero Martín JC, Pereira MP, Serrano Sanz S, et al. Dysfunctional oxidative phosphorylation shunts branched-chain amino acid catabolism onto lipogenesis in skeletal muscle. EMBO J 2020;39(14):e103812. [PMID: 32488939 DOI: 10.15252/embj.2019103812]
- Shah SH, Crosslin DR, Haynes CS, Nelson S, Turer CB, et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia 2012;55(2):321-30. [PMID: 22065088 DOI: 10.1007/s00125-011-2356-5]
- Holeček M. The role of skeletal muscle in the pathogenesis of altered concentrations of branched-chain amino acids (Valine, Leucine, and Isoleucine) in liver cirrhosis, diabetes, and other diseases. Physiol Res 2021;70:293-305. [PMID: 33982576 DOI: 10.33549/physiolres.934648]
- Lynch CJ, Adams SH. Branched-chain amino acids in metabolic signalling and insulin resistance. Nat Rev Endocrinol 2014;10(12):723. [PMID: 25287287 DOI: 10.1038/nrendo.2014.171]
- McGarrah RW, White PJ. Branched-chain amino acids in cardiovascular disease. Nat Rev Cardiol 2023;20(2):77-89. [PMID: 36064969 DOI: 10.1038/s41569-022-00760-3]
- Zhao S, Zhou L, Wang Q, Cao JH, Chen Y, et al. Elevated branched-chain amino acid promotes atherosclerosis progression by enhancing mitochondrial-to-nuclear H2O2-disulfide HMGB1 in macrophages. Redox Biol 2023;62:102696. [PMID: 37058999 DOI: 10.1016/j.redox.2023.102696]
- Roe DS, Roe CR, Brivet M, Sweetman L. Evidence for a short-chain carnitine-acylcarnitine translocase in mitochondria specifically related to the metabolism of branched-chain amino acids. Mol Genet Metab 2000;69(1):69-75. [PMID: 10655160 DOI: 10.1006/mgme.1999.2950]
- Violante S, IJlst L, Ruiter J, Koster J, Van Lenthe H, et al. Substrate specificity of human carnitine acetyltransferase: implications for fatty acid and branched-chain amino acid metabolism. Biochim Biophys Acta 2013;1832(6):773-9. [PMID: 23485643 DOI: 10.1016/j.bbadis.2013.02.012]
- McCalley S, Pirman D, Clasquin M, Johnson K, Jin S, et al. Metabolic analysis reveals evidence for branched chain amino acid catabolism crosstalk and the potential for improved treatment of organic acidurias. Mol Genet Metab 2019;128(1–2):57-61. [PMID: 31133529 DOI: 10.1016/j.ymgme.2019.05.008]
- Wang J, Liu Y, Lian K, Shentu X, Fang J, et al. BCAA catabolic defect alters glucose metabolism in lean mice. Front Physiol 2019;10:1140. [PMID: 31551816 DOI: 10.3389/fphys.2019.01140]
- Sun L, Liang L, Gao X, Zhang H, Yao P, et al. Early prediction of developing type 2 diabetes by plasma acylcarnitines: a population-based study. Diabetes Care 2016;39(9):1563-70. [PMID: 27388475 DOI: 10.2337/dc16-0232]
- Mihalik SJ, Goodpaster BH, Kelley DE, Chace DH, Vockley J, et al. Increased levels of plasma acylcarnitines in obesity and type 2 diabetes and identification of a marker of glucolipotoxicity. Obesity 2010;18(9):1695-700. [PMID: 20111019 DOI: 10.1038/oby.2009.510]
- Shi M, He J, Li C, Lu X, He WJ, et al. Metabolomics study of blood pressure salt-sensitivity and hypertension. Nutr Metab Cardiovasc Dis 2022;32(7):1681-92. [PMID: 35599090 DOI: 10.1016/j.numecd.2022.04.002]
- Kalhan SC, Guo L, Edmison J, Dasarathy S, McCullough AJ, et al. Plasma metabolomic profile in nonalcoholic fatty liver disease. Metabolism 2011;60(3):404-13. [PMID: 20423748 DOI: 10.1016/j.metabol.2010.03.006]
- Huang K, Li Z, He X, Dai J, Huang B, et al. Gut microbial co-metabolite 2-methylbutyrylcarnitine exacerbates thrombosis via binding to and activating integrin α2β1. Cell Metab 2024;36(3):598-616.e9. [PMID: 38401546 DOI: 10.1016/j.cmet.2024.01.014]
- Mohanta S, Yin C, Weber C, Hu D, Habenicht AJ. Aorta atherosclerosis lesion analysis in hyperlipidemic mice. Bio-Protoc 2016;6(11):e1833. [PMID: 27366759 DOI: 10.21769/bioprotoc.1833]
- Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb J Vasc Biol 1994;14(1):133-40. [PMID: 8274468 DOI: 10.1161/01.atv.14.1.133]
- Amorim N, McGovern E, Raposo A, Khatiwada S, Shen S, et al. Refining a protocol for faecal microbiota engraftment in animal models after successful antibiotic-induced gut decontamination. Front Med 2022;9:770017. [PMID: 35223890 DOI: 10.3389/fmed.2022.770017]
- Santos-Baez LS, Ginsberg HN. Hypertriglyceridemia—causes, significance, and approaches to therapy. Front Endocrinol 2020;11:616. [PMID: 32982991 DOI: 10.3389/fendo.2020.00616]
- Raposeiras-Roubin S, Rosselló X, Oliva B, Fernández-Friera L, Mendiguren JM, et al. Triglycerides and residual atherosclerotic risk. J Am Coll Cardiol 2021;77(24):3031-41. [PMID: 34140107 DOI: 10.1016/j.jacc.2021.04.059]
- Packard CJ, Boren J, Taskinen MR. Causes and consequences of hypertriglyceridemia. Front Endocrinol 2020;11:252. [PMID: 32477261 DOI: 10.3389/fendo.2020.00252]
- Deprince A, Haas JT, Staels B. Dysregulated lipid metabolism links NAFLD to cardiovascular disease. Mol Metab 2020;42:101092. [PMID: 33010471 DOI: 10.1016/j.molmet.2020.101092]
- Zhao J, Zhang Y, Wei F, Song J, Cao Z, et al. Triglyceride is an independent predictor of type 2 diabetes among middle-aged and older adults: a prospective study with 8-year follow-ups in two cohorts. J Transl Med 2019;17:403. [PMID: 31801571 DOI: 10.1186/s12967-019-02156-3]
- Tao LX, Yang K, Liu XT, Cao K, Zhu HP, et al. Longitudinal associations between triglycerides and metabolic syndrome Components in a Beijing adult population, 2007-2012. Int J Med Sci 2016;13(6):445-50. [PMID: 27279794 DOI: 10.7150/ijms.14256]
- Xu S, Ilyas I, Little PJ, Li H, Kamato D, et al. Endothelial dysfunction in atherosclerotic cardiovascular diseases and beyond: from mechanism to pharmacotherapies. Pharmacol Rev 2021;73(3):924-67. [PMID: 34088867 DOI: 10.1124/pharmrev.120.000096]
- Khatana C, Saini NK, Chakrabarti S, Saini V, Sharma A, et al. Mechanistic insights into the oxidized low-density lipoprotein-induced atherosclerosis. Oxid Med Cell Longev 2020;2020:5245308. [PMID: 33014272 DOI: 10.1155/2020/5245308]
- Owens GK, Kumar MS, Wamhoff BR. Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol Rev 2004;84(3):767-801. [PMID: 15269336 DOI: 10.1152/physrev.00041.2003]
- Badimon L, Vilahur G. Thrombosis formation on atherosclerotic lesions and plaque rupture. J Intern Med 2014;276(6):618-32. [PMID: 25156650 DOI: 10.1111/joim.12296]