MicroRNAs in Liver Fibrosis: Unraveling Intercellular Crosstalk and the Regulatory Functions of MicroRNA-223
1Department of Biochemistry, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand
2Center of Excellence in Hepatitis and Liver Cancer, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand
3Medical Science Program, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand
*Correspondence to: Chaiyaboot Ariyachet, Department of Biochemistry, Faculty of Medicine, Chulalongkorn University, Bangkok 10330, Thailand. E-mail: Chaiyaboot.A@chula.ac.th, cariyach@gmail.com
Received: March 24 2025; Revised: May 17 2025; Accepted: May 28 2025; Published Online: June 18 2025.
Cite this paper:
Phetkong C, Ariyachet C. MicroRNAs in Liver Fibrosis: Unraveling Intercellular Crosstalk and the Regulatory Functions of MicroRNA-223. BIO Integration 2025; 6: 1–6.
DOI: 10.15212/bioi-2025-0060. Available at: https://bio-integration.org/
Download citation
© 2025 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Liver fibrosis is caused by excessive extracellular matrix accumulation, which in turn is driven by activation of hepatic stellate cells. MicroRNAs (miRNAs), a class of small non-coding RNAs, are increasingly recognized as crucial regulators of gene expression in liver pathology, including fibrosis, by influencing intercellular crosstalk. Among these miRNAs, microRNA-223 (miR-223) has emerged as a key regulator of fibrosis, beyond its originally recognized role in innate immunity. By traveling between liver cell types via exosomes, miR-223 modulates inflammation and fibrosis via Hedgehog, platelet-derived growth factor (PDGF), and transforming growth factor-β signaling, and additionally dampens hepatic stellate cell activation by repressing glioma-associated oncogene homolog 2, PDGF receptor β, α-smooth muscle actin, and autophagy related 7, thereby influencing mechanotransduction and autophagy. However, numerous studies have reported opposite results, in which miR-223 promotes fibrosis under certain conditions, thus underscoring the roles of cellular environments, expression levels, and competing RNA networks. This Commentary synthesizes the current, sometimes contradictory, evidence; outlines how context shapes miR-223’s dual actions; and surveys the development of therapeutic strategies, including miRNA mimics, nanoparticle formulations, and extracellular-vesicle delivery, including major challenges in tissue targeting, cargo stability, and long-term safety. These insights together highlight miR-223 as a complex but intriguing target for antifibrotic treatment.
Keywords
Extracellular vesicles, hepatic stellate cell, liver fibrosis, microRNA-223, miR-223, non-coding RNA.
Introduction
Liver fibrosis, a common result of chronic liver injury such as hepatitis, metabolic dysfunction-associated steatotic liver disease (MASLD), cirrhosis, and hepatocellular carcinoma, disrupts liver function [1]. In liver fibrosis, the characteristic excessive extracellular matrix deposition is driven primarily by hepatic stellate cell (HSC) activation. After injury, HSCs transform into myofibroblasts and overproduce type I collagen. The lack of effective antifibrotic therapies highlights the need to better understand HSC activation for new treatment strategies.
Whereas earlier studies have emphasized protein-coding pathways such as TGF-β/Smad and Wnt/β-catenin in HSC activation, non-coding RNAs (ncRNAs), including microRNAs (miRNAs), have also been found to play key roles [2]. Indeed, recent years have seen a surge in research highlighting the critical involvement of miRNAs in the pathogenesis and progression of liver fibrosis. Numerous miRNAs have been found to be dysregulated in fibrotic livers, thereby influencing key cellular processes such as inflammation, oxidative stress, apoptosis, and critically HSC activation and excessive extracellular matrix production [2]. This growing body of evidence underscores the potential of miRNAs to serve as both diagnostic biomarkers and therapeutic targets. These 18–22 nucleotide RNAs regulate gene expression post-transcriptionally by targeting messenger RNAs. Among the many miRNAs implicated in liver fibrosis, microRNA-223 (miR-223) has notably garnered considerable attention, because its dysregulation, particularly downregulation, is consistently observed during HSC activation [3], thereby suggesting a key role in controlling HSC fibrotic phenotypes. This Commentary summarizes the mechanistic functions of miR-223 in HSC activation and fibrosis, and discusses its therapeutic potential.
Biosynthesis and function of miR-223 in the normal liver
The conserved miR-223 gene on the X chromosome is regulated by myeloid transcription factors (PU.1, C/EBP, and NFI-A) [4]. This miRNA, first identified in myeloid cells, controls granulocyte differentiation and promotes anti-inflammatory M2 macrophage polarization. Despite its low abundance in many cell types, miR-223 is transferred via extracellular vesicles (EVs). These vesicles carry messenger RNAs, ncRNAs, and proteins, thus mediating intercellular communication. In the liver, hepatocytes absorb neutrophil-derived miR-223–enriched EVs through low-density lipoprotein receptor and apolipoprotein E interaction [3]. In hepatocytes, miR-223 regulates drug and cholesterol metabolism and may enhance chromosomal stability and consequently decrease hepatocellular carcinoma risk [4]. Its roles in HSCs, cholangiocytes, and endothelial cells remain unclear and merit further study for therapeutic development.
MiR-223 in liver injury and fibrosis
Liver fibrosis is driven by tissue damage and inflammation, in which cytokines (e.g., TGF-β, TNF-α, and IL-6) from neutrophils and macrophages promote HSC activation [1]. miR-223 acts as an antifibrotic factor, given that miR-223−/− mice have been found to exhibit elevated liver injury and fibrosis. Mechanistically, miR-223 suppresses neutrophil overactivation in acetaminophen (APAP)-induced liver injury [5]. APAP triggers mitochondrial DNA release, thereby activating Toll-like receptor 9 in neutrophils and inducing nuclear factor-κB (NF-κB)-mediated inflammation. miR-223, upregulated by NF-κB, silences IKK-α and consequently terminates inflammation via a negative feedback loop. Consistently, miR-223-knockout mice show enhanced susceptibility to APAP-induced injury and elevated liver inflammation. Likewise, in alcohol-induced liver injury, miR-223 resolves inflammation by inhibiting the IL-6/p47phox pathway and directly targeting IL-6 [6].
Beyond its role in neutrophil function, miR-223 influences the broader inflammatory microenvironment contributing to liver fibrosis. In a recent study, in the absence of miR-223, an increase in macrophages in the liver has been found to further exacerbate inflammation and accelerate fibrosis [7]. Macrophages contribute to liver fibrosis by secreting TGF-β1 and other pro-fibrotic cytokines. This dysregulation of immune cell recruitment underscores the critical roles of miR-223 in modulating immune cell accumulation and suppressing excessive inflammatory responses, thereby limiting fibrotic progression in the liver.
Although miR-223 is abundant in neutrophils, it is packaged into EVs and delivered to other liver cells, where it aids in resolving inflammation and fibrosis after hepatic injury [8, 9]. In a mouse model of chronic liver injury, neutrophil-derived miR-223 suppresses NLRP3 inflammasomes in hepatic macrophages, thus limiting cytokine release and promoting M2 polarization. Neutrophil ablation prolongs M1 presence, thereby worsening inflammation, whereas delivery of an miR-223 analog has opposite effects. These findings challenge the notion that neutrophils are solely detrimental in liver inflammation and fibrosis.
Beyond macrophages, hepatocytes also take up neutrophil-derived miR-223 via EVs, and consequently contribute to the resolution of inflammation and fibrosis. In a high-fat diet mouse model of MASLD, fatty acids and IL-6 induce miR-223 biogenesis in neutrophils and promote the delivery of miR-223-containing EVs to hepatocytes [10]. Consequently, myeloid-specific knockout of IL-6 receptor α decreases miR-223 expression in hepatocytes and subsequently facilitates the activation of miR-223 target genes, including transcriptional activator with PDZ-binding motif (TAZ). Upregulation of TAZ promotes fibrosis through the Yes-associated protein (YAP)/TAZ mechanosensing pathway, which induces hepatocyte secretion of Indian hedgehog (IHH) and activation of hedgehog signaling in HSCs.
MiR-223 in HSC activation
Recent work has highlighted the roles of miR-223 in immune cells and hepatocytes in fibrosis. Emerging evidence indicates that miR-223 reaches HSCs via EVs and modulates their activation (Figure 1 and Table 1). Strong miR-223 downregulation in activated human HSCs suggests an anti-fibrotic role [11]. Despite low intrinsic levels of miR-223 in HSCs, exosomal miR-223 from neutrophils and natural killer (NK) cells modulates HSC activation. Glioma-associated oncogene homolog 2 (Gli2) is a direct target of miR-223 in HSCs [12]. Gli2, a GLI-family transcription factor, undergoes nuclear translocation after hedgehog signaling and subsequently promotes fibrogenesis. miR-223-mediated silencing of Gli2 may block IHH from hepatocytes and subsequently limit HSC activation. Because miR-223 also impairs TAZ-driven IHH induction in hepatocytes, neutrophil miR-223 restricts fibrosis by targeting multiple genes in the TAZ–IHH–Gli2 axis linking neutrophils, hepatocytes, and HSCs. YAP/TAZ inhibition in myofibroblasts also attenuates liver fibrosis. Therefore, neutrophil miR-223 dampens this pathway in hepatocytes and HSCs, thereby attenuating the fibrogenic response. Nevertheless, whether miR-223 directly regulates TAZ in hepatocytes and HSCs remains to be experimentally tested.
Figure 1 Roles of miR-223 in suppressing HSC activation. Neutrophil-derived miR-223 can be transferred via EVs to hepatocytes and HSCs. In hepatocytes, miR-223 silences expression of TAZ, thus decreasing secretion of IHH, a ligand that stimulates hedgehog signalling in HSCs via PTCH binding. In HSCs, miR-223 suppresses the transcription factor Gli2, a mediator of hedgehog signaling. Furthermore, miR-223 inhibits PDGF and TLR4 signaling, ATG7-mediated autophagy, and cytoskeletal remodeling by α-SMA. These processes are all known to promote HSC activation. HSC: hepatic stellate cell; EV: extracellular vesicle; TAZ: transcriptional activator with PDZ-binding motif; IHH: Indian hedgehog; PTCH: patched-1 receptor; PDGF: platelet-derived growth factor; TLR4: Toll-like receptor 4; α-SMA: α-smooth muscle actin; ATG7: autophagy related 7. Created with BioRender.
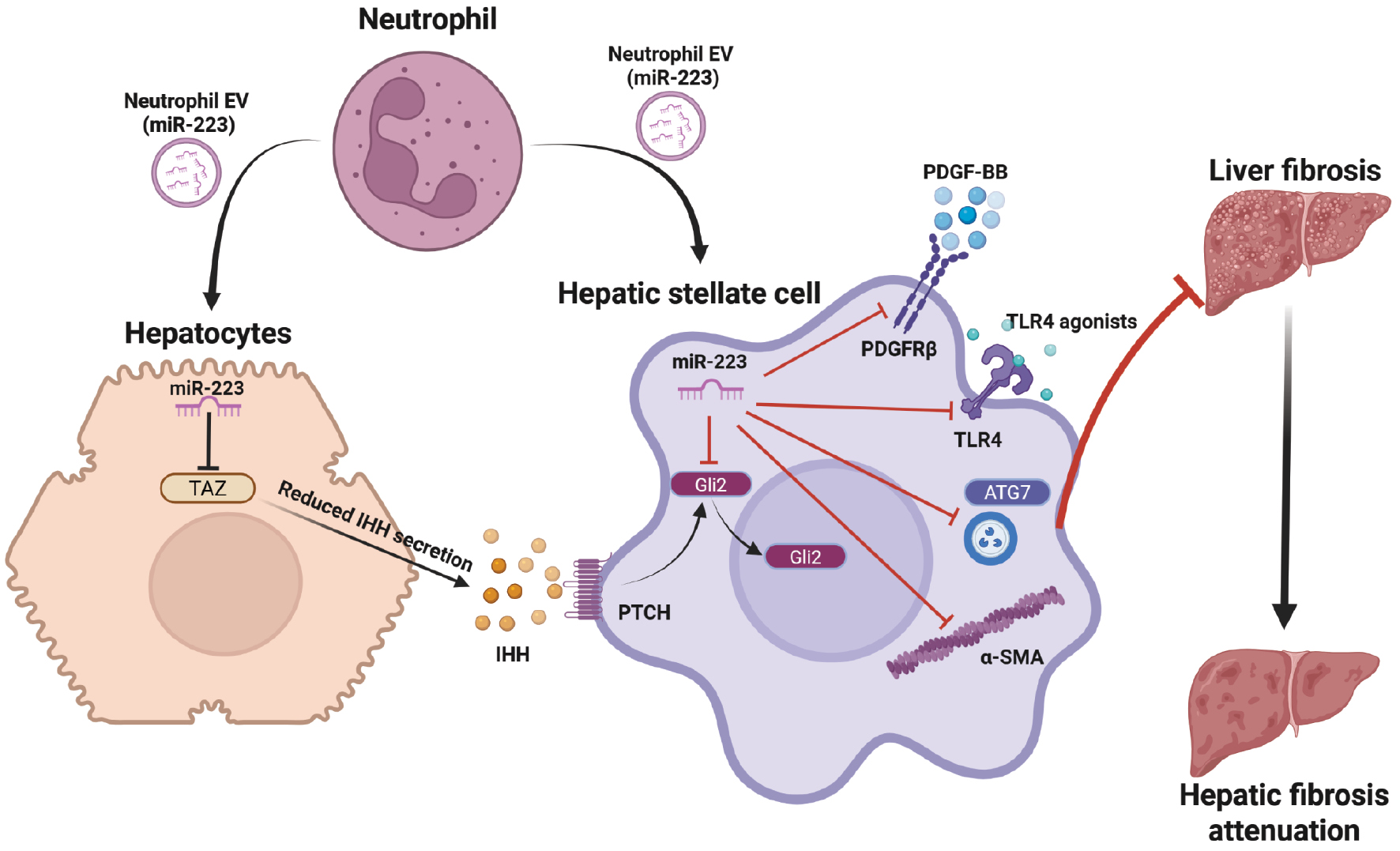
Table 1 Roles of anti-fibrotic miR-223 in various hepatic cell types
| Cell Type | Target | Validation Type | Study Model | Mechanism | Reference |
|---|---|---|---|---|---|
| Neutrophil | IKK-α | In vivo | Mouse model of APAP-induced drug injury | MiR-223 inhibits NF-κB signaling, thereby terminating inflammatory and fibrogenic responses | [5] |
| Neutrophil | IL-6 | In vivo | Mouse model of alcohol-induced drug injury | MiR-223 inhibits production of IL-6, thereby blocking neutrophil infiltration and generation of reactive oxygen species | [6] |
| Macrophage and T cell | PD-1/PD-L1 | In vivo | Mouse models of DEN and CCl4-induced liver injury | MiR-223 downregulates immunosuppressive (PD-1/PD-L1) signals, thereby decreasing fibrosis and the infiltration of PD-1+ T cells and PD-L1+ macrophages | [7] |
| Macrophage | NLRP3 | In vivo | Mouse models of CCl4-induced liver injury and MCD diet | MiR-223 inhibits formation of NLRP3 inflammasomes and stimulates macrophage differentiation toward M2 phenotypes | [8, 9] |
| Hepatocyte | TAZ | In vivo | Mouse high-fat diet models of MASLD | MiR-223 inhibits YAP/TAZ signaling, a pathway that induces IHH expression and subsequently activates Hedgehog signaling in HSCs | [10] |
| HSC | Gli2 | In vivo | Mouse models of CCl4-induced liver injury | MiR-223 decreases Gli2 expression and hence impairs Hedgehog signaling stimulated by hepatocyte IHH | [12] |
| HSC | PDGFRβ | In vivo | Mouse models of CCl4-induced liver injury | MiR-223 negatively regulates PDGF signaling, thereby promoting activation phenotypes of HSCs | [12] |
| HSC | α-SMA | In vitro | Primary human HSCs and hepatic organoids | MiR-223 inhibits α-SMA expression and impairs mechanotransduction of HSCs | [13] |
| HSC | ATG7 | In vitro | Human LX-2 HSC cell line | MiR-223 downregulates ATG7 expression and interrupts autophagy | [14] |
| HSC | TLR4 | In vitro | Human LX-2 HSC cell line | MiR-223 impairs TLR4 signaling, thereby promoting HSC activation | [15] |
IKK-α: IκB kinase alpha; APAP: acetaminophen; IL-6: interleukin-6; NLRP3: NOD-, LRR- and pyrin domain-containing protein 3; CCl4: carbon tetrachoride; DEN: diethylnitrosamine: PD-1: programmed cell death protein 1; PD-L1: programmed death ligand-1; MCD: methionine-choline deficient; HSC: hepatic stellate cell; TAZ: transcriptional activator with PDZ-binding motif; YAP: yes-associated protein; IHH: Indian hedgehog; PDGF: platelet-derived growth factor; α-SMA: α-smooth muscle actin; ATG7: autophagy related 7; TLR4: Toll-like receptor 4.
Beyond the TAZ–IHH–Gli2 pathway, miR-223 may also regulate platelet-derived growth factor (PDGF) signaling, a pathway that promotes HSC activation, proliferation, and migration [12]. Bioinformatic analysis has predicted that miR-223 binds the regulatory region of the PDGFRB transcript. Overexpression of miR-223 in HSCs decreases PDGFRβ levels and inhibits PDGF-BB-induced signaling. However, although this result is consistent with bioinformatic predictions, direct binding of miR-223 to the PDGFRB transcript in HSCs has not been experimentally validated. Nevertheless, their direct interaction has been confirmed in vascular smooth muscle cells [12].
Furthermore, miR-223 exhibits an antifibrotic role by regulating cytoskeletal activity [13]. After HSC activation, α-smooth muscle actin (α-SMA) upregulation drives cytoskeletal rearrangement and generates mechanical signals that promote fibrogenesis. miR-223 directly suppresses α-SMA expression in HSCs, and its overexpression decreases cell migration, invasion, and contractility by impairing myofibroblast mechanotransduction.
Beyond neutrophils, NK cells transfer miR-223 to HSCs via EVs, thereby suppressing HSC activation [14]. NK cell-derived miR-223 directly suppresses autophagy-related 7 (ATG7), a central autophagy regulator that recycles dysfunctional components. Therefore, exosomal miR-223-mediated ATG7 downregulation curbs autophagy in HSCs and weakens TGF-β-driven activation. However, some studies have indicated that autophagy can reverse fibrosis, thus underscoring the complexity of the miR-223-autophagy-HSC axis and the need for further investigation.
Beyond transfer via EVs, miR-223 is regulated by ncRNAs such as circular RNAs (circRNAs). Circular RNA PWWP2A (circ-PWWP2A) acts as a competing endogenous RNA that enhances HSC activation and proliferation via sequestering miR-223; elevating Toll-like receptor 4; and driving fibrogenic, inflammatory, and anti-apoptotic responses [15]. Another competing endogenous RNA, hsa_circ_0070963, has an opposite effect: its overexpression inhibits fibrosis, because miR-223 normally suppresses LEM domain-containing 3 (LEMD3), a negative regulator of TGF-β signaling. By sponging miR-223, hsa_circ_0070963 restores LEMD3 and dampens TGF-β-induced HSC activation [16]. Notably, downregulation of hsa_circ_0070963 during liver fibrosis increases miR-223-3p availability and consequently suppresses LEMD3 expression. Subsequently, a decrease in LEMD3 attenuates its inhibitory effect on TGF-β signaling, ultimately promoting HSC activation and fibrogenesis. Functional analyses have shown that miR-223 enhances fibrotic responses and partly reverses the anti-fibrotic effects of hsa_circ_0070963. Collectively, these findings suggest that hsa_circ_0070963 protects against fibrosis by sequestering miR-223, restoring LEMD3 function, and inhibiting TGF-β-driven HSC activation. Given these contradictory findings, further studies are needed to clarify the regulatory interactions between circRNAs and miR-223 in HSC activation.
MiR-223 as a therapeutic target for liver fibrosis
As previously discussed, miR-223 plays an antifibrotic role by regulating multiple biological functions across hepatic cell types, and subsequently mitigating inflammation and fibrosis. Enhancing miR-223 bioavailability in the damaged liver is therefore a promising therapeutic strategy to suppress fibrogenesis. miRNA-based therapies have been developed for several liver diseases [17]. Lipid-based nanoparticles delivering miR-223 have been found to decrease inflammation and fibrosis in murine models of acute and chronic hepatic injury [8, 9]. Similarly, adenovirus-mediated miR-223 (Ad-miR-223) significantly attenuates fibrosis and alleviates inflammation by suppressing PD-1+ CD8+ T cells and PD-L1+ macrophages (IBA1+ cells), thus highlighting its roles in immune modulation and fibrotic regulation [7]. Given the broad antifibrotic effects of miR-223 across hepatic cell types, targeted delivery of this miRNA to immune cells, hepatocytes, and HSCs might offer a more effective therapeutic approach for liver fibrosis and hepatic inflammation.
However, EV-based therapies still face several hurdles. Because naked miRNAs are rapidly degraded by serum nucleases, chemical modification or encapsulation is essential, yet these steps can alter biological activity. Other challenges include improving exosome yield and purity, achieving precise tissue-specific delivery without off-target gene silencing, maintaining cargo stability, and generating comprehensive long-term biodistribution and toxicology data. Nevertheless, the platform remains highly promising; a recent study has demonstrated the therapeutic potential of miR-223-loaded EVs from adipose-derived mesenchymal stem cells in mitigating liver fibrosis [18]. These adipose-derived mesenchymal stem cell-EV miR-223 particles are readily taken up by hepatocytes, and subsequently decrease lipid accumulation and fibrotic responses in a mouse model of MASLD. Similarly, EVs loaded with miR-223 from bone marrow stem cells alleviate inflammation and cell death in autoimmune hepatitis, probably via miR-223-mediated suppression of NLRP3 and caspase-1 [19].
The clinical translation of miR-223-based therapies will require further progress in delivery optimization, dose selection, and long-term safety evaluation. Prior experience from miRNA-based interventions, including those halted because of immune-related toxicity, highlights the importance of careful risk assessment and therapeutic monitoring. Although manufacturing and specificity challenges remain, continued advances in RNA therapeutics and delivery platforms offer a promising foundation for the clinical development of well-characterized miRNA candidates [20]. Further research focusing on optimizing EV engineering for targeted delivery and enhanced loading, as well as establishing safe and precise delivery methods, will be crucial for translating these findings into clinical applications.
Limitations, controversies, and future perspectives
In conclusion, miR-223 has emerged as a critical player in liver physiology and pathophysiology, particularly in the context of liver fibrosis. Although its primary role was initially associated with immune regulation, its ability to be delivered via EVs and exert therapeutic effects on hepatic cells is promising. However, several key questions remain to be answered, pertaining to clinically relevant dosing ranges, optimal treatment durations, and the long-term safety of sustained miR-223 modulation [4]. Delivery platforms that can direct miR-223 to hepatocytes, HSCs, and inflammatory subsets with minimal off-target silencing also require rigorous optimization. Another clearly identified gap is the absence of harmonized fibrosis endpoints and robust dose-response data, both of which are essential to guide future experimental design (Table 2). Filling these gaps will rely on research that combines ex vivo human liver slices, organoids from patients, and humanized mouse models to understand how specific cells respond, as well as multi-omics profiling to delineate the related networks, and pharmacokinetic and immunogenicity data to establish safety limits. The knowledge gained will enable biomarker-guided, dose-escalation trials in well-characterized fibrosis cohorts, wherein circulating miR-223 levels and non-invasive fibrosis scores can be used both for patient stratification and real-time monitoring of therapeutic responses. Existing discrepancies in research findings underscore the need for larger animal and human-relevant models to clarify the context-dependent actions of miR-223. Furthermore, leveraging advanced technologies such as single-cell sequencing and bioinformatics would enable the construction of detailed gene regulatory networks and offer novel insights into the pathogenic mechanisms of liver fibrosis. Ultimately, these efforts should pave the way to the development of targeted and effective miR-223-based antifibrotic treatments.
Table 2 Knowledge gaps and future research directions for miR-223 in liver fibrosis
| Area of focus | Current Gap | Proposed Research Direction |
|---|---|---|
| Cell specificity | Dual antifibrotic and profibrotic roles depending on cell type | Elucidate cell-specific functions through single-cell and spatial transcriptomic profiling |
| ncRNA interaction | Unclear crosstalk with other classes of ncRNAs and unclear direct gene targets | Investigate interactions with circRNAs and lncRNAs; experimentally validate direct miR-223 targets |
| Target network | Insufficient understanding of downstream signaling and miRNA pleiotropy | Integrate multi-omics approaches to define gene regulatory networks; functionally confirm miR-223 targets |
| Clinical evidence | Limited data from human tissues and models | Use human liver organoids, ex vivo human liver slices, and patient-derived cells for translational validation |
| Therapeutic window | Undefined optimal dosing regimen and treatment duration | Perform dose-ranging, PK, and persistence studies to optimize therapeutic parameters |
| Delivery strategy | Inefficient targeting, poor stability, and risk of off-target effects | Engineer EVs or nanoparticles with specificity to hepatocytes or HSCs and enhanced delivery stability |
| Drug approval process | Lack of preclinical data to support clinical translation | Implement GLP-compliant toxicology and initiate IND-enabling studies, per FDA/EMA regulatory standards |
ncRNAs: non-coding RNAs; circRNAs: circular RNAs; lncRNAs: long non-coding RNAs; miRNA: microRNA; EVs: extracellular vesicles; HSC: hepatic stellate cell; PK: pharmacokinetics; GLP: Good Laboratory Practice; IND: Investigational New Drug; FDA/EMA: U.S. Food and Drug Administration/European Medicines Agency.
Author contributions
Conceptualization, C.A.; original draft preparation, C.P. and C.A.; review and editing, C.P., and C.A.
Funding
No funding or sponsorship was received for this study.
Acknowledgements
None.
Conflicts of interest
The authors declare that there are no conflicts of interest.
References
- Kisseleva T, Brenner D. Molecular and cellular mechanisms of liver fibrosis and its regression. Nat Rev Gastroenterol Hepatol 2021;18(3):151-66. [PMID: 33128017 DOI: 10.1038/s41575-020-00372-7]
- Ezhilarasan D. MicroRNA interplay between hepatic stellate cell quiescence and activation. Eur J Pharmacol 2020;885:173507. [PMID: 32858048 DOI: 10.1016/j.ejphar.2020.173507]
- Avila MA. MicroRNA-223: a key regulator of liver tumour microenvironment. Gut 2023;72(10):1811-12. [PMID: 36792357 DOI: 10.1136/gutjnl-2022-329322]
- Ye D, Zhang T, Lou G, Liu Y. Role of miR-223 in the pathophysiology of liver diseases. Exp Mol Med 2018;50(9):1-12. [PMID: 30258086 DOI: 10.1038/s12276-018-0153-7]
- He Y, Feng D, Li M, Gao Y, Ramirez T, et al. Hepatic mitochondrial DNA/Toll-like receptor 9/MicroRNA-223 forms a negative feedback loop to limit neutrophil overactivation and acetaminophen hepatotoxicity in mice. Hepatology 2017;66(1):220-34. [PMID: 28295449 DOI: 10.1002/hep.29153]
- Li M, He Y, Zhou Z, Ramirez T, Gao Y, et al. MicroRNA-223 ameliorates alcoholic liver injury by inhibiting the IL-6-p47phox-oxidative stress pathway in neutrophils. Gut 2017;66(4):705-15. [PMID: 27679493 DOI: 10.1136/gutjnl-2016-311861]
- Fu Y, Mackowiak B, Feng D, Lu H, Guan Y, et al. MicroRNA-223 attenuates hepatocarcinogenesis by blocking hypoxia-driven angiogenesis and immunosuppression. Gut 2023;72(10):1942-58. [PMID: 36593103 DOI: 10.1136/gutjnl-2022-327924]
- Jimenez Calvente C, Del Pilar H, Tameda M, Johnson CD, Feldstein AE. MicroRNA 223 3p negatively regulates the NLRP3 inflammasome in acute and chronic liver injury. Mol Ther 2020;28(2):653-63. [PMID: 31585800 DOI: 10.1016/j.ymthe.2019.09.013]
- Calvente CJ, Tameda M, Johnson CD, Del Pilar H, Lin YC, et al. Neutrophils contribute to spontaneous resolution of liver inflammation and fibrosis via microRNA-223. J Clin Invest 2019;129(10):4091-109. [PMID: 31295147 DOI: 10.1172/JCI122258]
- Hou X, Yin S, Ren R, Liu S, Yong L, et al. Myeloid-cell-specific IL-6 signaling promotes MicroRNA-223-enriched exosome production to attenuate NAFLD-associated fibrosis. Hepatology 2021;74(1):116-32. [PMID: 33236445 DOI: 10.1002/hep.31658]
- Coll M, El Taghdouini A, Perea L, Mannaerts I, Vila-Casadesús M, et al. Integrative miRNA and gene expression profiling analysis of human quiescent hepatic stellate cells. Sci Rep. 2015;5(1):1-14. [DOI: 10.1038/srep11549]
- Wang X, Seo W, Park SH, Fu Y, Hwang S, et al. MicroRNA-223 restricts liver fibrosis by inhibiting the TAZ-IHH-GLI2 and PDGF signaling pathways via the crosstalk of multiple liver cell types. Int J Biol Sci 2021;17(4):1153-67. [PMID: 33867837 DOI: 10.7150/ijbs.58365]
- Ariyachet C, Chuaypen N, Kaewsapsak P, Chantaravisoot N, Jindatip D, et al. MicroRNA-223 suppresses human hepatic stellate cell activation partly via regulating the actin cytoskeleton and alleviates fibrosis in organoid models of liver injury. Int J Mol Sci 2022;23(16):9380. [PMID: 36012644 DOI: 10.3390/ijms23169380]
- Wang L, Wang Y, Quan J. Exosomal miR-223 derived from natural killer cells inhibits hepatic stellate cell activation by suppressing autophagy. Mol Med. 2020;26(1):81. [PMID: 32873229 DOI: 10.1186/s10020-020-00207-w]
- Liu W, Feng R, Li X, Li D, Zhai W. TGF-β- and lipopolysaccharide-induced upregulation of circular RNA PWWP2A promotes hepatic fibrosis via sponging miR-203 and miR-223. Aging 2019;11(21):9569-80. [PMID: 31719209 DOI: 10.18632/aging.102405]
- Ji D, Chen GF, Wang JC, Ji SH, Wu XW, et al. Hsa_circ_0070963 inhibits liver fibrosis via regulation of miR-223-3p and LEMD3. Aging 2020;12(2):1643-55. [PMID: 32003753 DOI: 10.18632/aging.102705]
- Wang X, He Y, Mackowiak B, Gao B. MicroRNAs as regulators, biomarkers and therapeutic targets in liver diseases. Gut 2021;70(4):784-95. [PMID: 33127832 DOI: 10.1136/gutjnl-2020-322526]
- Niu Q, Wang T, Wang Z, Wang F, Huang D, et al. Adipose-derived mesenchymal stem cell-secreted extracellular vesicles alleviate non-alcoholic fatty liver disease via delivering miR-223-3p. Adipocyte 2022;11(1):572-87. [PMID: 36093813 DOI: 10.1080/21623945.2022.2098583]
- Chen L, Lu FB, Chen DZ, Wu JL, Hu ED, et al. BMSCs-derived miR-223-containing exosomes contribute to liver protection in experimental autoimmune hepatitis. Mol Immunol. 2018;93:38-46. [PMID: 29145157 DOI: 10.1016/j.molimm.2017.11.008]
- Seyhan AA. Trials and tribulations of MicroRNA therapeutics. Int J Mol Sci. 2024;25(3):1469. [PMID: 38338746 DOI: 10.3390/ijms25031469]