Advances in Gene Therapy for Neurologic Disorders: An Overview
Pallavi Chand1,*![]() , K. Trideva Sastri2, Ashish Singh Chauhan1, Souvik Chakraborty3 and Vikash Jakhmola4
, K. Trideva Sastri2, Ashish Singh Chauhan1, Souvik Chakraborty3 and Vikash Jakhmola4
1Department of Pharmaceutics, Uttaranchal Institute of Pharmaceutical Sciences, Uttaranchal University, Dehradun 248007, Uttarakhand, India
2Department of Pharmaceutics, JSS College of Pharmacy, JSS Academy of Higher Education & Research, Mysuru, Karnataka, India
3Department of Pharmaceutics, Eminent College of Pharmaceutical Technology, Barbaria, Jagannathpur, Barasat, WB, India
4Department of Pharmaceutical Chemistry, Uttaranchal Institute of Pharmaceutical Sciences, Uttaranchal University, Dehradun 248007, Uttarakhand, India
*Correspondence to: Dr. Pallavi Chand, Uttaranchal Institute of Pharmaceutical Sciences, Uttaranchal University, Dehradun 248007, Uttarakhand, India, Tel: +91-8979302352. E-mail: pallavichand1990@gmail.com
Received: July 28 2024; Revised: October 7 2024; Accepted: October 17 2024; Published Online: February 5 2025
Cite this paper:
Chand P, Sastri KT, Chauhan AS et al. Advances in Gene Therapy for Neurologic Disorders: An Overview. BIO Integration 2025; 6: 1–17.
DOI: 10.15212/bioi-2024-0060. Available at: https://bio-integration.org/
Download citation
© 2025 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Neurologic disorders currently affect approximately 100 million people worldwide. Neurologic disorders most often occur due to inherent genetic mutations, which lead to numerous types of functional disruptions in nervous system development. Neurologic disease-related events, such as genetic and epigenetic changes, cause inflammatory processes in the area which may enhance the disease cycle. Gene therapy has progressed to a compelling therapeutic approach for various neurodegenerative disorders. Several efforts to enhance gene therapy rely on discovering novel vectors, recent curative targets, and the dependability of transgenic delivery paths. These viral and non-viral vectors techniques are carefully screened through preclinical and clinical levels and eventually render patients with effective therapies. This review addresses gene therapy developments and obstacles for neurodegenerative diseases and discusses emerging strategies, goals, and prospects.
Keywords
Adeno-associated viruses, gene therapy, lipid-based vectors, neurologic disorders, non-viral and viral vectors.
Introduction
Gene therapy has made substantial advances in the treatment of neurodegenerative illnesses, such as Alzheimer’s disease (AD), progressive supranuclear palsy, Huntington’s disease (HD), Parkinson’s disease (PD), ataxia, motor neuron disease, multiple system atrophy, and etc. Discerning pathogenetic pathways with a deeper understanding of these disorders has driven numerous advances in converging essential technologies, including discovering newer beneficial targets and vectors [1]. Broad knowledge has led to ground-breaking targeting of single and complex gene etiologies by various genomic treatments of the core reasons regarding neurodegenerative disorders. Gene therapy facilitates prolonged and enduring therapeutic potentials specifically for localized actions in organs, like the eye and cochlea, gene therapy exhibits prolific results in the central nervous system (CNS). Gene therapy produces an alternate treatment for agents that fail to penetrate numerous physiologic barriers [2–4]. In addition, precise genetic targets unyielding to conventional therapy are theoretically pliable by gene therapy, gene silencing capable of handling function mutation advantages, and gene overexpression to manage function mutation losses. Figure 1 shows the basic workings of gene therapy.
Figure 1 Basic gene therapy.
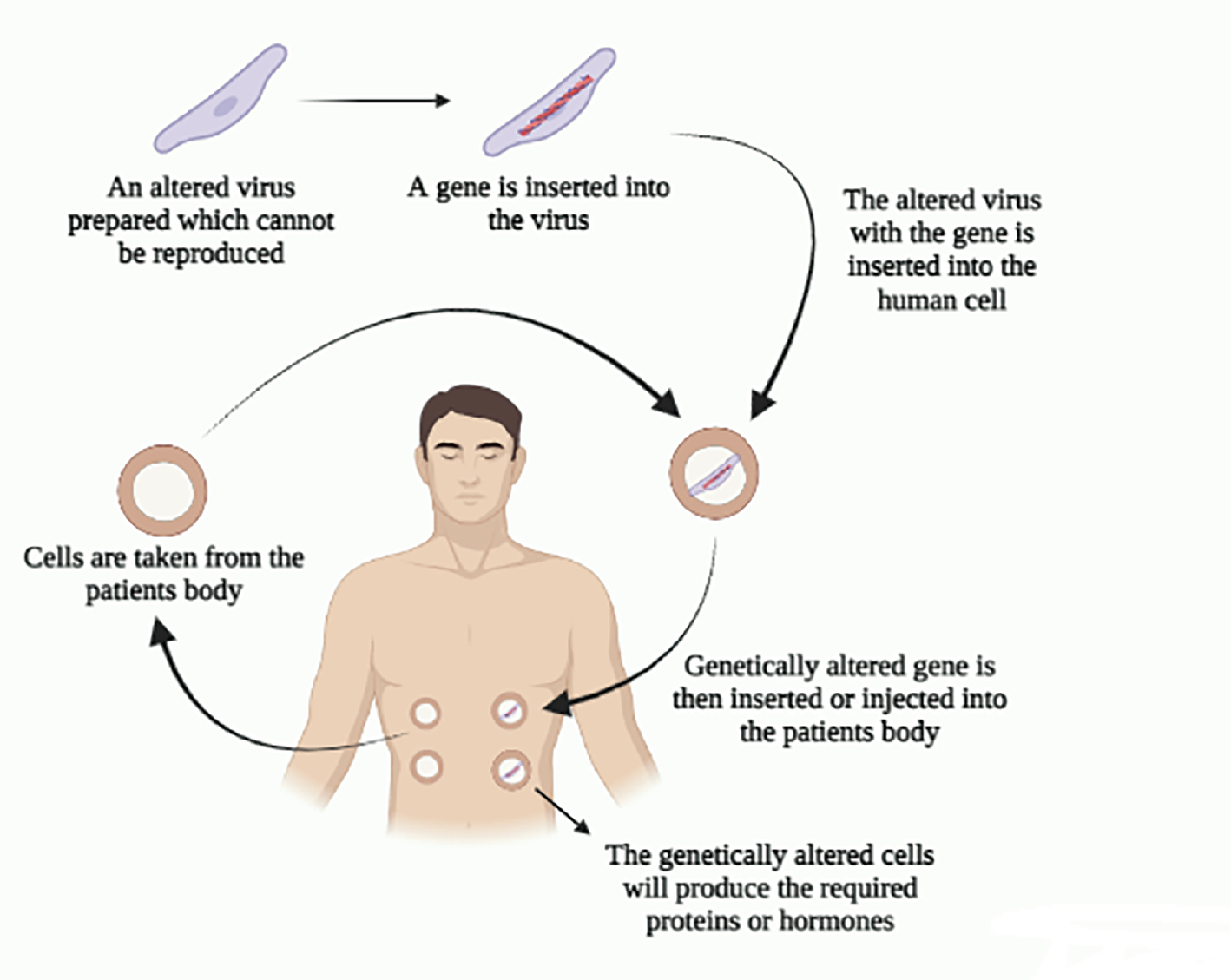
The problem to be solved in gene therapy for neurologic disorders is the need for effective treatments that can address the underlying genetic causes of these conditions. Current treatments often have limited efficacy and are plagued by side effects. Gene therapy offers a promising approach by directly targeting the genetic mutations responsible for these disorders [2–4]. However, significant challenges remain, including the development of safe and efficient delivery methods, the ability to regulate transgene expression, and the need for more effective animal models to guide human clinical trials. The manuscript highlights the significance of gene therapy in treating neurological disorders. Gene therapy offers a promising approach to addressing the underlying genetic causes of these conditions, which are often incurable by conventional methods [2–4]. The manuscript emphasizes the potential of gene therapy to restore missing gene function, overcome the blood-brain barrier (BBB), and provide a humane and cost-effective alternative to traditional treatments. It also discusses the challenges and future directions in gene therapy, including the delivery methods and the effective animal models to guide human clinical trials [2–4]. Our study aimed to understand the mechanisms and molecular basis of cognitive and motor impairment in neurologic disorders, exploring gene therapy as a potential treatment approach. It discusses the current techniques, challenges, and recent developments in gene therapy for neurologic disorders, including PD, AD, and HD. Existing knowledge on gene therapy for neurologic disorders includes advances in adeno-associated virus (AAV) vectors, conditional expression strategies, and preclinical research for various conditions. However, challenges persist in optimizing safety and efficacy, and more effective animal models are needed to guide human clinical trials [2–4].
Current gene therapy approaches face limitations, such as the need for improved AAV capsids, regulation of transgene expression, and the lack of effective animal models. Additionally, the BBB and immune responses pose significant challenges for gene therapy delivery and efficacy [2–4]. Our study will benefit individuals suffering from neurologic disorders, such as PD, HD, and AD. Its implications include the potential for gene therapy to become a viable treatment option for these conditions, offering a humane and cost-effective alternative to conventional treatments.
Our study contributes to the advancement of gene therapy for neurologic disorders by the following:
- Exploring the potential of gene therapy in treating various neurologic conditions;
- Discussing the current techniques, challenges, and recent developments in gene therapy;
- Highlighting the need for improved AAV capsids, regulation of transgene expression, and effective animal models; and
- Providing insights into the mechanisms underlying and molecular basis for cognitive and motor impairment in neurologic disorders.
Viral and non-viral-based gene therapy
Human and animal vectors efficaciously regulate transgenes that manifest therapeutic antibodies, small interfering (si)RNA, and mRNA in unhealthy cells. AAVs are among the most frequently used vectors in neurodegenerative conditions. In addition to several astrocyte concerns, oligodendrocytes are successfully targeted with a broad range of capsids [5–8]. Further evolution of non-viral delivery systems and advances in AAVs and gene therapies have portrayed safe profiles and extensive transgene expression. Moreover, recent experimental findings revealed significant outcomes for several diseases, such as neurodegenerative disorders [9–12].
Viral vectors
Vectors based on AAVs have been functional for neurodegenerative disorders, specifically in clinical phases. Serotypes belonging to AAVs are the primary determining factor of several critical features of active gene therapy based on AAVs, with tissue tropism, biodistribution, and exposure to in vivo-induced antibody neutralization [10–12]. Understanding how various serotypes transport genes to their target tissues for vector administration is crucial in building a successful and reliable approach to gene therapy. Over 100 AAV variations have been documented, including 13 serotypes (AAV1-13) derived from non-human primates and humans. AAV2, known for its relative safety and sustained expression in neurons, has been extensively researched in many clinical studies and is considered a potential vector for gene therapy targeting neurodegenerative diseases [13, 14].
It has been recommended that AAV2-NGF abides by intracerebral administration and displays a significant therapeutic effect on cognitive impairment in Alzheimer-related dementia. Remarkably, AAV4 tends to transfect ependymal cells that account for the epithelial neuroblast lining and lateral ventricles after processing into cerebral ventricles. The BBB is the most significant barrier in transmitting the utmost vectors to the CNS. AAV9 and AAVrh10 effectively overcome this obstacle [15, 16].
It has been reported that a newly developed AAV capsid (AAV-PHPB) can transduce >50% of neurons and astrocytes, which delivers AAV genomes 40 times higher than other capsids to CNS post-intravenous injection, illustrating the essential worth of capsid engineering [17]. However, extensive cell-type scrutiny of various AAV capsid libraries has shown that AAV capsids accompany extensive tropisms. Adenovirus (Adv) is an icosahedral capsid virus approximately 70–100 nm in size [18]. Adv is incompetent in transfecting its gene into the genome, following relative ephemeral transgene expression in an admirable protection profile. The innate immune stimulus to Adv limits the scope of the Adv therapeutic effect on CNS gene therapy [19]. Although specific studies were carried out using Adv for treating neurodegenerative disorders, the studies reveal Adv borne with few side effects.
In contrast to Adv and AAV capsids, retroviruses and lentiviruses (LVs) potentially incorporate complete DNA via reverse transcription into the host genome, providing more stable and prolonged in vivo transgenes. These insertions must be regulated under rigorous conditions to prevent genotoxicity and insertional mutagenesis. To date, there has been one significant clinical trial using a LV vector that proficiently delivers greater PD DNA loads. The records showed that ProSavin, a gene therapy dependent on LV vectors expected to restore the development of dopamine, enhance motor activity and establish safety in all patients with advanced PD. Further research into gene therapy of neurodegenerative diseases induced by Adv and LVs is urgently needed, despite the existing inadequate clinical evidence [20].
Non-viral vectors
Clinical trials have studied AAVs, Adv, LV, and retrovirus vectors to deliver therapeutic genes. However, several drawbacks are associated with these vectors, with large tropism, limited loading potential, difficulties related to vector manufacture, and inflammatory responses in the host. Gene therapies with non-viral vectors may avoid many drawbacks, especially safety-related issues [21, 22]. Therefore, although few of these strategies have been used clinically, the exploitation of new vectors, predominantly nanoparticles, and lipid-carrier systems, is highly significant. Non-viral transmission vectors are classified into lipidic polymeric vectors dependent on the carrier material composition. The most frequently used non-viral gene carriers are vectors that are lipid-dependent. Cholesterol, dioleoylphosphatidylethanolamine (DOPE) and 1,2-distearoyl-sn-glycero-3-phosphorylethanolamine (DSPE) are neutral lipids that have been utilized as the “helper lipids” (liposomal components) to improve system stability and transfection capability. Cationic lipids are highly suitable for gene therapy, such as 1,2-dioleoyl-3-trimethylammoniumpropane (DOTAP) and N-[1-(2,3-dioleyloxy)propyl]-N,N,N-trimethylammonium (DOTMA). These lipids contain three main domains (linking groups, hydrophobic tails, and cationic caps). The primary constraints of cationic lipids are the suboptimal pharmacokinetic biodistribution caused by non-specific binding and rapid clearance, as well as cytotoxicity [23–27]. Cationic lipids with suitable pKas have successfully resolved these drawbacks. In addition to lipidoids, exosomes, and magnetic nanoparticles have been established as potential gene carriers for neurodegenerative disorders [28–30].
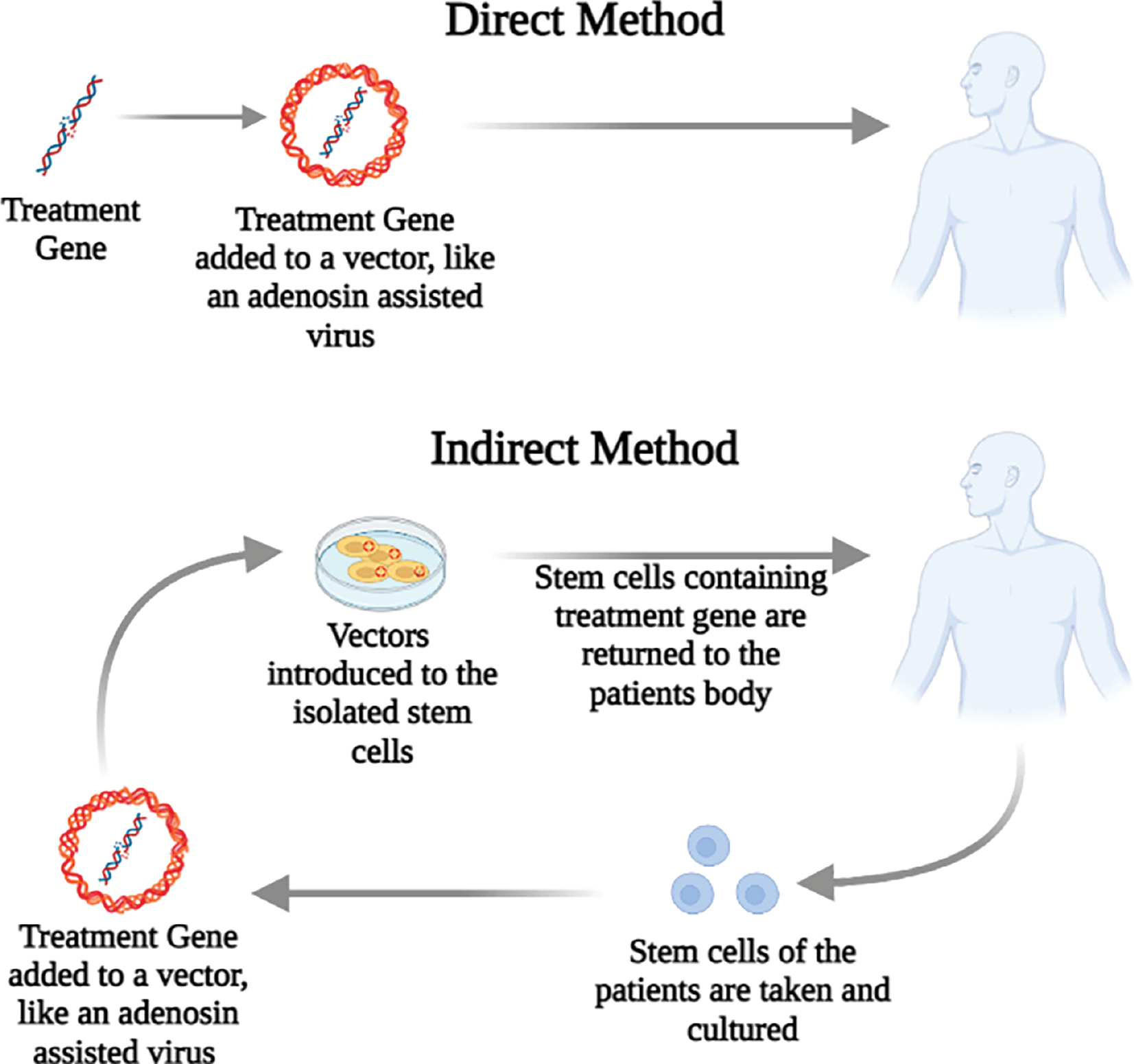
Recent research has demonstrated that Fe3O4 magnetic nanoparticles, which are covered with N-isopropyl acrylamide derivatives and oleic acid molecules and carry shRNA-α-syn have a significant therapeutic effect in alleviating PD in mice. Cationic polymers deliver a different type of non-viral vector, which is enormously effective in gene therapy because of its ability to escape endosomes and lysosomes due to the sponge-proton effect, well-globular architecture, and enormous chemical diversity [31, 32]. Further acumen into the connection amid the structural function of gene delivery material and more thorough knowledge of essential factors that inhibit effective gene delivery probably improves clinical care for neurodegenerative disorders. Figure 2 describes the types of indirect and direct delivery methods used in gene therapy, which includes acquiring genetic material that alters endogenous or exogenous gene expression [33]. Gene therapy methods can be utilized to contain a transgene to improve cellular functions. Generally, a sequence is given that encodes the wild-type human isomers of an enzyme. This therapeutic technique can be used in patients with a defective non-functional gene to have a working gene. This practice is applied to produce proteins (growth factors) for enhancement of cell survival [34].
Figure 2 Methods of gene therapy administration, including both direct and indirect approaches.
Multiple sclerosis
Multiple sclerosis (MS) is one of the foremost progressive impairments in young adults that is triggered by CNS inflammation, demyelination, and axonal failure. MS faces a rigorous hindrance in designing novel therapies because the etiology remains uncertain, the pathogenesis is only partly understood, and the entire cycle is multifocal, recurrent, and occurs across anatomic boundaries, making it challenging to supply potentially therapeutic molecules. Therefore, gene therapy is a practical option for ensuring sustained and site-specific therapy delivery. Recent developments in understanding the immunopathologic mechanisms that contribute to inflammation of the CNS, and the development of new gene therapy techniques, such as RNA-interference, are increasingly leading to a wide range of intervention possibilities [35].
While the precise pathogenesis of MS has not been established, the highly significant pathogenic traits have been suggested for CD4+ T-cell-mediated autoimmunity. The effector helper T cells responsible for resolving the pathophysiology underlying MS and experimental autoimmune encephalomyelitis (EAE) were the T-helper type 1 (Th1) cells that express interferon-gamma (IFN-γ). This study demonstrated that the T-helper subset, namely Th17 cells, has a pivotal function in chronic and autoimmune inflammation. Additional studies have shown that inhibiting IL-17 in mice may ameliorate several autoimmune disorders, such as EAE. Furthermore, brain tissue damage in individuals with MS has been shown to include T cells that produce IL-17 [36, 37].
Mammalian non-coding micro-RNAs (miRNAs) have been proposed to participate in the development of the immune system. We observed a strong link between the level of miR-155 expression and the severity of the illness humans with MS and mice with EAE. There are low numbers of Th1 and Th17 cells that downregulate miR-155. This link resulted in a weak form of EAE. In contrast, increasing miR-155 leads to more Th1 and Th17 cells, which makes EAE worse. MiR-155 encouraged the growth of Th17 and Th1 cells, which cause inflammation. These results showed that miR-155 makes people more susceptible to EAE by changing how T cells react to inflammation. MiR-155 could also serve as a new target for treating MS [38].
A review of the current small non-coding (snc)RNAs in MS showed significant differences between the results expected from heterogeneity at the level of cohorts, samples, and procedures used. However, the findings suggested that dysregulation of miRNAs intracellularly and circulating offer prospects of miRNAs for alleviating MS. MiRNAs suggest control cell activities and reveal essential roles in the immune and CNS tissue of MS patients. SncRNA and miRNA studies can enhance our knowledge of phenotypic variation linked to developing the disease, subtype, or clinical parameters and elucidate mechanisms underpinning MS. In addition, sncRNA and miRNA profiling may provide robust biomarkers for enhanced diagnosis, prognosis, and response prediction. The often-charged use of RNS-based treatments could be a promising method for personalized medicine in the long term. Future work in translating results from sncRNA research with clinical practice for patients with MS is instrumental [39].
The movement of inflammatory immune cells, such as Th17 and Th1, into the CNS has been shown to contribute to the demyelination of the sheath of the neurons, thus inducing neuro-inflammation and neurodegeneration. Long non-coding RNAs (lncRNAs) are non-protein-coding transcripts longer than 200 nucleotides (nts). LncRNAs have a crucial role in regulating large genes and pathways involved in cell growth, specialization, cell death, and cell development, especially in the differentiation of immune cells and the activation of the adaptive immune system. LncRNAs are linked with other lncRNAs Based on MS studies, lncRNAs have lower expression and more specificity in tissues, cell types, and developmental stages compared to protein-coding genes. This finding suggests that lncRNAs may have a role in the progression of specific diseases. Dysregulation is believed to be a significant factor in the development of several autoimmune disorders (AIDs) according to multiple studies [39–42].
Linc-MAF-4
Linc-MAF-4 was initially identified in 2015 as a regulator of CD4+ T cell development through control of MAF transcription and chromatin modifier transcription. Zhang et al. conducted a microarray investigation to examine the expression of lncRNAs in peripheral blood mononuclear cells (PBMCs) of six MS patients and five healthy controls. The study revealed that linc-MAF-4 is significantly increased in MS patients [43]. Subsequently, the levels of linc-MAF-4 were artificially increased and decreased in CD4+ T cells obtained from an additional 28 MS patients to demonstrate variations in the characteristics and activities of different subsets of CD4+ T cells [43, 44]. Linc-MAF-4 was shown to enhance the differentiation of Th1 cells, while inhibiting the differentiation of Th2 cells. Linc-MAF-4 was also shown to be a crucial factor in the differentiation of Th1 cells and the production of IFN-γ. Furthermore, linc-MAF-4 induced the activation of CD4+ T cells in individuals with MS without impacting proliferation. Subsequently, a correlation study was conducted that revealed the level of linc-MAF-4 expression was linked to the yearly frequency of relapses in patients with MS. These findings indicated that linc-MAF-4 has a role in the development of MS, primarily by regulating the encephalitogenic activity of Th1 cells. Linc-MAF-4 governs the process of CD4+ T cell development by regulating the transcription of chromatin modifiers. Linc-MAF-4 exhibits a notable increase in expression in individuals with MS, hence facilitating the development of Th1 cells and diminishing the differentiation of Th2 cells. Additionally, linc-MAF-4 induces activation of CD4+ T cells without impacting proliferation. The expression of linc-MAF-4 has a correlation with the yearly recurrence rate in patients with MS, suggesting involvement in the development of MS by regulating Th1 cells [44].
HOTAIR
Antisense transcript intervened RNA (HOTAIR), an lncRNA a 2148 nts in length, is reproduced from the HoxC gene group into chromosome 12 in an antisense fashion. Numerous studies have established HOTAIR pathogenesis in a variety of malignancies and inflammatory diseases [45]. The HOTAIR role in MS pathogenesis has recently been investigated concerning vitamin D levels using in vivo, in vitro, and silico findings. The findings found that higher levels of HOTAIR disease have been detected in MS patients than controls before treating vitamin D [46]. A trend toward low levels of HOTAIR with treatment has been shown with vitamin D, which also reached levels close to those of controls. In addition, the regulatory function of HOTAIR was examined in vivo and the monocytic THP-1 cells of humans in MS-like EAE mice [47]. HOTAIR expression differs in the CNS of EAE mice; specifically, HOTAIR is decreased in the cortex and spinal cord and elevated in the cerebellum. However, for the cell-based functions of HOTAIR, no changes in HOTAIR expression were found in the in vitro THP-1 cells analysis, vitamin D treatment, or inflammation induction. Overall, our findings suggest that HOTAIR is probably implicated in the development of MS and pathways associated with HOTAIR will be modified based on the impact on gene expression, inflammation, and vitamin D. Further investigation is needed to explore this association in greater detail [48, 49].
1700040D17Rik IL23R-CHR
An extracellular domain, truncated IL23R, can block in vitro cellular signals in-between IL-23 endogenous IL23R and Th17 [50]. Furthermore, in vivo analysis in EAE mice showed IL23R-CHR exhibits efficacy for treating EAE by suppressing CNS inflammation and generating pro-inflammatory cytokines. To recognize the pharmacologic activity of the IL23R-CHR, the profiling lncRNA-1700040D17Rik between the normal and EAE mice is designated lncRNAs [51]. After IL23RCHR treatment, a mouse intergenic lncRNA (1700040D17Rik) near the RORγt gene, is significantly increased in EAE mice. When 1700040D17Rik is over-expressed, the level of RORγt expression is decreased significantly and the development of Th17 cells is also reduced in vitro in the mice. The above results indicated the association between the biological role of 1700040D17Rik and the separation of Th17 cells due to control of RORγt expression. Considering that 1700040D17Rik has a potential role in the pathogenesis of EAE, the IL23R-CHR effects may be attributable to the response balancing of the immune response [52].
Huntington’s disease
HD is a severe neurologic disorder that is inherited in an autosomal dominant manner. HD often begins in early adulthood and is characterized by a range of symptoms affecting mental health, movement, and cognitive function [53]. The condition is attributed to a mono-allelic mutation that involves an abnormal increase in the number of CAG trinucleotide repeats in the HTT gene, which is responsible for encoding the huntingtin protein (HTT) [54]. The occurrence of this condition exhibits regional variation, with rates ranging from 0.40 per 100,000 individuals in Asian communities to 13.7 per 100,000 individuals in some Western groups [55]. The diagnosis of HD has traditionally been based on the presence of a motor phenotype that seems normal clinically either through a family history suggestive of the disease or by confirmation of a specific genetic mutation [56]. HD is caused by a malfunctioning repetition of three nucleotides (CAG) on the shorter section of the fourth chromosome, specifically in exon 1 of the HTT gene [57]. HD is an ideal candidate for genetic therapy due to its high penetrance as a monogenic illness with an extended pre-manifestation period and a single mutation that results in the production of a single rogue protein with cellular consequences [58].
In a study conducted by Sava and colleagues, a transgenic YAC128 mouse model of HD was utilized to administer enriched chitosan-based nanoparticles containing anti-HTT siRNA directly from the nose to the brain. A set of chitosan-based nanoparticles (NPs) was developed to enclose anti-HTT siRNA and safeguard anti-HTT siRNA from degradation during transportation to the intended destination. Factors contributing to the enhancement of effective anti-HTT siRNA nano-carrier production were discovered and evaluated in a YAC128 mouse model of HD. The study conducted by Sava et al. investigated the use of chitosan-based NPs loaded with anti-HTT siRNA for delivering the treatment directly from the nose to the brain in a YAC128 mouse model of HD. The main objectives were to safeguard the siRNA from degradation and determine the key parameters necessary for the effective administration of the anti-HTT siRNA nano-carriers [59].
In a gene therapy study, Kacher et al. demonstrated that targeting the delivery of CYP46A1, the enzyme responsible for regulating cholesterol breakdown in the brain, has a durable neuroprotective effect in HD. This approach effectively counteracts various harmful effects caused by the mutated huntingtin protein [60].
Spronck et al. administered AAV5-miHTT by injection into the striatum of Q175 HD mice. The administration of AAV5-miHTT resulted in a decrease in the HTT protein that was dependent on the dosage and persisted over time. This reduction effectively suppressed the formation of mutant HTT aggregates in both the striatum and cortex. Zinc finger protein transcription factors (ZFP-TFs) inhibit the repetition and specifically reduce the pathogenic CAG of mHTT as a therapeutic strategy [61]. Experiments employing fibroblasts and neurons obtained from patients showed that ZFP-TFs effectively suppress >99% of alleles that cause HD over a wide range of doses, while still preserving >86% of normal gene expression. Other genes that include CAG are only slightly affected. Zinc finger protein transcription factors (ZFP-TFs) given by viral means exhibit sustained activity and are readily absorbed by HD neurons in a laboratory setting for >100 days. Furthermore, ZFP-TFs remain active in the mouse brain for a minimum of 9 months [62]. Verma et al. investigated the effects of glucose-dependent insulinotropic polypeptide (GIP) receptor agonists on the neurobehavioral consequences of quinolinic acid-induced HD in rats. Administering 300 nmol/4 μL of quinolinic acid to the rat striatum bilaterally caused the rat to exhibit abnormal locomotive activity, impaired motor control, compromised neuromuscular coordination, and short-term episodic memory deficits. Treatment with a continuous GIP receptor agonist, capable of penetrating the brain, for 14 days [63].
Pfister et al. employed AAV9 to specifically transport an engineered miRNA that targets exon 48 of the human HTT mRNA to the striatum of HD sheep. This delivery was achieved by utilizing two different promoters or CβA. At both 1 and 6 months following the injection, the treatment resulted in a reduction of human mutant (m) HTT mRNA and a 50%–80% decrease in protein levels in the striatum. Measurable silencing occurred in both the caudate and putamen. The amounts of endogenous sheep HTT protein have remained constant. There was no significant decrease in the number of neurons tagged with DARPP32 or NeuN 6 months after diagnosis. Microglia expressing Iba1 were at normal levels [64].
Yang et al. discovered that by using CRISPR/Cas9-mediated inactivation, the production of mutant huntingtin (mHTT) was permanently decreased in the striatum of HD140Q-knock-in mice. This method efficiently eliminated the accumulation of HTT aggregates and reduced early neuropathology. Manipulating the expression of mHTT in the striatal neuronal cells of adult HD140Q-knock-in mice did not have an impact on survival but it did alleviate motor impairments. The results suggest that non-allele-specific CRISPR/Cas9-mediated gene editing can be used to efficiently and permanently eliminate polyglutamine expansion-induced neuronal toxicity in the adult brain [65]. Yang et al. also devised gene-silencing strategies that concentrate on the development of cassette-optimized artificial miRNAs (miHTTs). The original approach successfully achieved full suppression of both the wild-type and mtHTT genes by specifically targeting exon 1. In this strategy, allele-specific silencing was achieved by specifically targeting the heterozygous single-nucleotide polymorphism (SNP) rs362331 in exon 50 or 67 rs362307, which is connected to mtHTT. The miHTT expression cassette was enhanced by including target anti-HTT sequences into 10 primiRNA scaffolds and evaluating effectiveness in reducing HTT levels in vitro. This evaluation specifically focused on allele selectivity, passenger strand function, and processing patterns. Furthermore, three scaffolds encoding miH12, which specifically targets exon one, were incorporated into an adeno-associated viral serotype 5 (AAV5) vector [66]. The efficacy in reducing HTT expression and controlling pre-miHTT was assessed in the Hu128/21 HD mouse model, which is a transgenic mouse model that has been humanized [73].
Alzheimer’s disease
AD is a prevalent neurodegenerative disorder linked to aging. This condition is marked by the deterioration and irreversible loss of brain cells and connections, leading to a substantial decline in memory and cognitive abilities. Possible explanations for the development of AD involve oxidative stress, the buildup of Aβ plaques, disruption of the BBB, excessive movement of plasma proteins (e.g., fibrinogen into empty spaces in the brain), abnormal interactions between Aβ and fibrinogen, abnormalities in cholinergic neurotransmission, natural contamination, excessive phosphorylation of tau proteins, genetic factors (e.g., mutations in the amyloid precursor protein and presenilin genes), toxic effects of mutated RNA transcripts, and abnormal responses of microglia and the immune system [67].
Current AD treatments are aimed at alleviating symptoms by inhibiting acetyl-cholinesterase, which breaks down acetylcholine, or by blocking abnormal N-methyl-D-aspartic acid (NMDA) receptors, which are involved in glutamate signaling. Unfortunately, there are currently no available treatments that can alter the progression of the disease. While genetic proof, such as mutations in Aβ precursor protein (APP), presenilin 1 (PS1), PS2, and the enzymes that cleave Aβ from APP, is significant for the contribution of Aβ in the pathogenesis of AD, multiple shortcomings involving Aβ immunotherapies pose inquiries whether single Aβ-reducing treatment is sufficient for efficient treatment of AD. Aβ immunotherapies underline the need for target selection abroad [68]. Higher levels of Aβ are correlated through both family AD (FAD) and sporadic AD (SAD), suggesting that it will be helpful to reduce Aβ levels. Several clinical trials have been conducted over recent decades aimed at lowering levels of Aβ by immunotherapy with antibodies guided to other types of Aβ or secretase inhibitors (BACE1 and γ-secretase) with a focus on the generation of Aβ from APP. Although the immunotherapy approach has not been adopted in the clinic setting, the consequences on cognitive function have been minimal. The cause for the repeated failure can be attributed to cohort design, intervention period, or poor targeted engagement performance because of negligible passage of antibodies through the BBB. Therefore, immunotherapies will be costly and inaccessible to a substantial portion of the human population [69, 70]. An alternative is anti-Aβ aggregation compounds, in which the expression of recombinant proteins can form. The secretase inhibitor problems involve the secretases that target other proteins than APP. The γ-secretase modulator, which reduces the production of toxic Aβ42 and increases the production of the less toxic Aβ40, is a class of molecules that can substitute for complete inhibition of γ-secretase. Reducing APP rates will be another option. SiRNA mediates downregulation of APP by herpes simplex virus (HSV)-short hairpin RNA (shRNA) in APP overexpressing mice, which decreases Aβ pathology [71]. APP has various biological roles, the complete absence of which affects locomotive and cognitive dysfunctions [72]. Total deletion of APP is not possible, but rates of APP need to be evaluated with care. The use of AAV to deliver gene therapy targeting a specific proteolytic fragment of the APP, known as sAPPα, has shown potential neurotrophic effects. This treatment successfully restores spine density and improves working memory in mice with the APP/dE9 mutation [73].
BACE1
BACE1 reduces siRNA, cleaning Aβ from APP at the N-terminal, reducing pathology of Aβ, and decreasing behavioral deficiencies in an AD mouse model via LV discharge [74]. Targeted delivery of independent exosomes on a dendritic cell to reduce immunogenicity and effectively fill with BACE siRNA also decreases levels of BACE1. BACE1-affected mice exhibit hypomyelination and have elevated seizures, which is a significant factor for gene therapy and established BACE1 will require dosage with proper care [75].
PPAR gamma coactivator 1 alpha
PPAR gamma (PPARγ), a transcription factor that controls the metabolism of fatty acids and glucose, may be activated by delivering viral genes with the help of PPARγ coactivator one alpha (PGC-1a). This activation leads to a decrease in the level of BACE1 expression, resulting in a reduction in Aγ deposition. This leads to improved spatial and memory recognition in a mouse model of AD. Additionally, abnormal gene expression of PGC-1a raises neprilysin expression, which leads to Aβ pathology. Remarkably, there is also an increase in brain-derived neurotrophic factor (BDNF), which could significantly preserve survival of pyramidal neurons. PPARγ has a vital role in elevating liver X receptors (LXRs), a group of nuclear receptors that oxidize to the form of cholesterol (oxysterols) and promote lipid transportation of genetic material, like ApoE and ABCA1 [76]. Administration of LXR in AD-induced mice enhances cognitive entrapment, although termination leads to extreme amyloid pathology [77, 78]. The downstream activity of LXR shows higher in vivo neurogenesis in the brain. Taken together, gene therapy based on PPARγ has many advantages, including reducing and clearing Aγ, improving the survival of neurons, lipid metabolism (except for carriers like ApoE4), and neurogenesis [79].
Anti-Aβ aggregation – BRICHOS
BRICHOS, which is comprised of “Bri2, chondromodulin-I, and SP-C,” is a strong chaperone that stops Aβ from forming amyloid. Bioinformatics methods have been used to explain what BRICHOS is and how BRICHOS works. For example, an intramolecular region that might work like a chaperone was used [80]. The domain was identified from Bri2 protein sequence alignments associated with family dementia (chondromodulin), which is correlated with C (pro-SP-C) prosurfactant protein and the chondrosarcoma and dementia families. The BRICHOS domain can be found in 12 correlated families of proteins and has been suggested to be elaborated in the pro-protein post-translational processing. While biosynthesis of the BRICHOS domain occurs in the regions of precursor protein with high β-sheet propensities, the formation and aggregation of amyloid are prevented [81, 82]. In vitro and in vivo analysis showed that human Bri2 and pro-SP-C, products of recombination of BRICHOS, suppress the toxic oligomer, which occurs due to preventing residues of Aβ40-42. Amyloid formation is significantly below the stoichiometric ratio [83]. Furthermore, BRICHOS viral expression in AD mouse models facilitates the homeostasis of proteins and decreases brain Aβ pathology. In vivo experiments of transgenic mice model and Drosophila melanogaster have also shown that the BRICHOS domain prevents neurotoxicity and cognitive loss induced by Aβ, which is correlated with decreased learning and memory induced ɣ-oscillations [84–86].
Furthermore, BRICHOS, which are derived from Bri2 protein, is more flexible and effective than pro-SP-C in preventing neurotoxicity mediated by Aβ42 and in fibrillation and aggregation pathways. The Bri2-derived BRICHOS is also highly effective in preventing locomotive abnormality caused by Aβ42 and reducing longevity [87]. In the end, regarding the above analysis, BRICHOS can be considered a possible way to manage AD, with further exploration requirements for its anti-amyloidogenic activity on Aβ.
Protein homeostasis – autophagy
Autophagy can be considered as a highly preserved catabolic pathway that controls the recycling and digestion of the damaged organelles, and also possibly toxic proteins, which include those that are implicated in TAR DNA-binding protein 43 (TDP-43), Aβ, polyglutamine-expanded HTT, tau, and α-synuclein, which are various neurodegenerative diseases [88]. The cytosolic cargo is sequestered in the preliminary phase of autophagy by an autophagosome (a de novo double-membrane vesicle). This mechanism is highly altered, including protein-linked autophagy (ATG), like Atg6 and Atg8, are well-known as Beclin 1 and LC3. Subsequently, goals for degradation are taken into consideration for autophagosomes through polyubiquitin-binding adaptor proteins, such as p62/SQSTM1 [89].
The buildup and clustering of improperly folded proteins within cells, which are broken down by the autophagy-lysosomal system, have been noted in different clinical manifestations of the majority of neurodegenerative illnesses [88]. In basal neuronal autophagy, neurodegenerative diseases decline with age. Atg5 or Atg7 are genetically deleted in mice caused by aggregopathic phenotype-like neurodegeneration, including the absence of unique pathogenic proteins [90]. In addition, abnormal changes in the Atg5 gene have been identified that prevent autophagy in congenital ataxia and cause developmental delay [91]. Frontotemporal dementia (FTD) and ALS are linked to mutations of P62/SQSTM1. PD is linked to mutations of Parkin and Pink1 [92, 93]. Additionally, AD patients perturb autophagy markers in post-mortem human brains. Ironically, degradation of autophagy mediates the secretion of various molecules implicated in neurodegenerative diseases. For example, in APP transgenic mice the deletion of Atg7 has been reported in the downfall of Aβ secretion and the formation of plaque [94].
Parkinson’s disease (PD)
PD is characterized by the degeneration of dopaminergic neurons in the basal ganglia, which is a result of complicated pathophysiologic processes. The involvement of excitatory glutamatergic and γ-aminobutyric acid (GABA) inhibition pathways is present in basal ganglia movement regulation [95]. Such changes contribute to the disinhibition of the output subthalamic nucleus (STN), which elevates the excitatory projections toward the substantia nigra pars reticularis (SNpr) and internal globus pallidus (GPi). The average effect is advancing the discharge of inhibition effect from SNpr and GPi to further basal ganglia nuclei, thalamus, and cortex, resulting in the peculiar characteristics of PD. Specific strategies, including high-frequency stereotactic lesion, pharmacologic silencing, and deep brain stimulation for the treatment of PD and targeting STN or GPi, have been applied to enhance motor control in PD [96–98].
Complex pathophysiology has been demonstrated in PD, which is not entirely understood and includes various brain structures and signaling pathways. Gene transfer is a relatively novel method of treating PD. This method is very flexible and multiple strategies for treating this disease have been explored [99]. The selection of a therapeutic target is based on three different approaches. Raising dopamine levels in the basal ganglia and transgenic enzymes are encoded by the cell signaling proteins present in the production or controlling the dopamine in the first and direct approach.
Enzymes that are involved in the metabolism of dopamine are GTP-cyclohydrolase-1 (GCH-1), tyrosine hydroxylase (TH), and aromatic amino acid decarboxylase (AADC). Experiments involving this method have shown successful results, whereas in humans, phase I/II studies are underway, having all three main enzymes (ProSavin) in an LV vector [100]. When the percentage of GABA preventing the excess activity of the STN is determined, the second approach can be considered for modulating the basal ganglia affected by PD. If successful, these strategies will provide symptomatic relief to the patients instead of altering the progression of the disease. Modified viruses (MVs) are used as the most common delivery vector in gene therapy. All the MVs will have incredibly limited viral DNA to bear agencies or genes of concern [101]. A minimal quantity of viral DNA from the MVs generates modified viral particles and does not express the viral protein.
In human studies various viruses are utilized as therapeutic gene therapy vectors, such as adenovirus, LV, AAV, and herpes virus [102–104]. Every virus has pros and cons with respect to therapeutic gene therapy. Pathogenic viruses, such as adenovirus and herpesvirus, can induce immune responses following transmission. Such vectors are widely used as an anti-tumor substance, which can potentially improve the treatment of cancer. LV and AAV have shown long-term expression for many years, despite being immunogenic [105]. These types of properties of viruses can be taken as suitable vectors for applications in other diseases.
The immunogenic viruses, LV and AAV, have been used in recent years for PD therapy. LV, a single-stranded RNA retrovirus, combines its genetic cargo with the present genome. LV has the ability for transgene or exogenous gene expression for an extended period. The combination of the host genome is a significant risk factor that disrupts an endogenous gene. Like LV, AAV is a single-stranded DNA virus that will not combine with the host genome. The virus introduces extra-chromosomal DNA, which does not disrupt the endogenous gene. Although the virus does not integrate into the host genomes, AAV still enables prolonged term gene expression owing to its stable extra-chromosomal construction. These two vectors have different carrying capacities of genetic materials. The carrying capacity of LVs is up to 8–9 kB, whereas AAV can accommodate <5 kB. Therefore, size limits may prohibit AAV use [106].
Improvement of dopamine synthesis
As previously discussed, L-DOPA has become a pharmacologic cornerstone for the management of PD. Dopamine levels are reduced in PD patients due to a deficiency in SNc dopaminergic neurons. L-DOPA has become an intermediate in the synthesis of the dopamine pathway [107]. The stories of dopamine levels can be raised by elevating the levels of L-DOPA. Keeping this in mind, some researchers used gene therapy to raise the rates of dopamine [108, 109]. The strategy involves provision of genes that help in coding the enzymes in the biosynthesis of the dopamine pathway. The most popular method is administering L-amino acid decarboxylase (AADC), which is aromatic, in the final stages of the synthesis of the dopamine pathway. The AADC transforms L-DOPA into dopamine. All groups that use this technique target the striatum/putamen for the transmission of the genes. Expression of AADC in the striatum/putamen can convert L-DOPA into dopamine at the location where AADC is typically produced [110].
Christine et al. recently used a skull compatible with MRI targeting systems to inject AAV-AADC bilaterally into the putamen [111]. Dissimilar to the studies mentioned above, which involved the conventional stereotactic method, the approach applied by the Voyager Therapeutics community provides a visualization of vector infusion because an MRI scanner is used for the construction of the injections of gadolinium contrast agent was mixed with the vector [112].
Delivery of trophic factors
The loss of dopaminergic neurons is an extremely critical factor in PD. Some groups tend to explore the delivery of growth factors of PD treatment. Gene expression with the growth factor increases neuron survival. Neurotrophic factor (GDNF) obtained from the glial cell line is an excessively investigated growth factor for treating PD. Initially, experiments were performed using a direct intracerebral infusion of the protein, GDNF. Preclinical studies in PD models of rodents and primates demonstrated excellent efficacy of direct GDNF protein infusion. Human phase 1 research has shown therapy effectiveness, which opens up further investigation [113]. However, randomized controlled trials of direct infusion of GDNF protein into the ventricular system were clinically unsuccessful [111, 114]. More experiments indicated that the trial failures might be due to the relatively small tissue distribution of the protein [114]. A recent intra-putamen randomized clinical trial involving GDNF focused on the restriction of tissue distribution through intermittent enhanced convection delivery via implanting a novel system. Scientists from the study inserted gadolinium into the system to assess the distribution of tissue. Randomization was used in patients with approximately 40% coverage of motor putamen MRI after administration through injection. Improvement in the motor putamen by UPDRS was noted after 40 weeks in the treatment and control groups. Although there was a tendency toward more significant change in the treatment community, the difference between the groups was not crucial statistically. The effect was apparent for the lower GDNF dose given safety concerns and an inadequate 40-week care/follow-up period, which is higher than predictable results in the control group for the possible placebo care of lesion effects and limited size of the sample. A subgroup of patients reported an increment of >10 points in the “off” UPDRS motor score in group therapy but not in the control group. This subgroup consisted primarily of patients aged between 60 and 75 years, with a moderate to severe stage of PD, typically having had the condition for over 10 years. Although the subgroup study cannot be revealed, the results can support further studies. In addition, an open-label extension study, which was not placebo-controlled, demonstrated continuation of clinical progress for 40 weeks of additional therapy, thereby endorsing a clinical trial with an endpoint of 80 weeks or <40 weeks as clinical trials related to gene therapy in Table 1, which shows a list of patents undergoing various gene therapies [115].
Table 1 Gene Therapy Approaches: Gene Targets and Delivery Methods [132]
| Identifier No. | Trial Type | Disease Type | Title | Phase | Status | Study Completion Date |
|---|---|---|---|---|---|---|
| NCT05040217 | Interventional (Clinical Trial) | Alzheimer’s disease | A clinical trial of AAV2-BDNF Gene therapy in early Alzheimer’s disease and mild cognitive impairment | Phase 1 | Recruiting | October 1, 2027a |
| NCT03634007 | Interventional (Clinical Trial) | Alzheimer’s disease | Gene therapy for APOE4 homozygote of Alzheimer’s disease | Phase 1 | Recruiting | December 2021a |
| NCT03306277 | Interventional (Clinical Trial) | Spinal muscular atrophy | Gene replacement therapy clinical trial for patients with spinal muscular atrophy Type 1 (STR1VE) | Phase III | Completed | November 12, 2019b |
| NCT00017940 | Interventional (Clinical Trial) | Alzheimer’s disease | Gene therapy for Alzheimer’s disease | Phase 1 | Completed | November 2003b |
| NCT05407636 | Interventional (Clinical Trial) | Age-related macular degeneration | Pivotal 2 Study of RGX-314 gene therapy in participants with NAMD | Phase 3 | Recruiting | December 2025a |
| NCT03634007 | Interventional (Clinical Trial) | Alzheimer’s disease | Gene therapy for APOE4 homozygote of Alzheimer’s disease | Phase 2 | Recruiting | November 2024a |
| NCT04167540 | Interventional (Clinical Trial) | Parkinson’s disease | GDNF Gene therapy for Parkinson’s disease | Phase I | Recruiting | June 2026a |
| NCT02122952 | Interventional (Clinical Trial) | Spinal muscular atrophy | Gene transfer clinical trial for spinal muscular atrophy Type 1 | Phase 1 | Completed | December 15, 2017b |
| NCT06444217 | Interventional (Clinical Trial) | Huntington’s disease | Gene therapy development and validation for Huntington’s disease fibro TG-HD (FibroTG-HD) | NA | Recruiting | July 2028a |
| NCT05040217 | Interventional (Clinical Trial) | Alzheimer’s disease, mild cognitive impairment | A clinical trial of AAV2-BDNF Gene therapy in early Alzheimer’s disease and mild cognitive impairment | Phase 1 | Recruiting | October 2027a |
aThe “estimated” study completion date is the date that the experts think the study will end. bThe moment that the last person in a clinical study was checked or given an intervention or treatment so that all the data could be collected.
Neuromodulation through gene therapy
The SNc is responsible for dopamine production, which is released in the striatum [116]. In a simplistic view of the circuit of basal ganglia, D1 neurons are excited in the presence of dopamine in the striatum on the “forward path.” As a result, the GPi is blocked by these neurons activating the cortex and disinhibiting the thalamus, which gives a “go signal” for motion. The “indirect pathway” for D2 neurons is activated by dopamine and disinhibits the external globus pallidus (GPe). GPe disinhibition contributes to the STN. Diminished STN activity results in reduced GP inactivity, which promotes movement by thalamus disinhibition. In the SN state, the dopaminergic neurons degenerate and dopamine levels decline in the PD state. This results in a reduction of immediate pathway excitation and reduces inhibition of the secondary pathway. The average effect reduces the excitation of the cortical thalamus, which predicts hyperactivity of the STN in the PD state. The STN hyperactivity is the presumed target of ablative case methods and DBS [117].
Gene transfer is a better method that enables almost any gene to be delivered. One researcher has developed a clever approach to gene therapy for modulating the activity of STN. Glutamatergic neurons are usually included in the STN. Glutamate is converted to GABA by glutamate decarboxylase (GAD). GAS expression is suggested in the STN to convert glutamate to GABA-type neurons and change STN production from the excitant to the repressive phase [118].
For the same reason, an AAV vector that carries GAD genetic material has been developed. Because the micro-dialysis test has been carried out, preclinical research results showed that the administration of AAV-GAD to the STN outcomes elevates the percentage of GABA in the production region of SN. The STN targeted structures have been inhibited and recorded in electro-physiologic tests. Although the GABA-transporter (vGaT) vesicular gene is not dispensed with GAD, the above-mentioned methods were successful, which may occur for the lower level expression of endogenous vGaT or for the induction of vGaT expression that occurs during GAD development. However, the micro-dialysis test is not tested directly in preclinical studies. Indeed, recent work has shown that single neurons release neurotransmitters diversely [119]. Evidence also suggests that neurons can give the vesicular glutamate transporters and vGaT in the STN. Despite the correct mechanism, the reported preclinical study showed an increase in the inhibition of STN targets [120].
Side effects and administration methods
While gene therapy offers promising avenues for treating neurologic disorders, it is essential to consider potential side effects. Side effects can include immune responses to the viral vectors used for gene delivery, which may lead to inflammation or other adverse reactions in the CNS. Additionally, the long-term effects of gene therapy are still being studied and there is a need for ongoing monitoring of patients to assess any delayed side effects that may arise from the treatment [121].
The method of administration is critical in determining the efficacy of gene therapy, especially in overcoming the BBB and the blood-cerebrospinal fluid barrier. Various delivery methods are being explored, including intracerebral administration, intrathecal injections, and direct injections into the spinal cord and brain [122]. Each method has advantages and challenges, and the choice of administration route can significantly impact the distribution and effectiveness of the therapeutic agents [123].
The BBB presents a significant challenge for gene therapy because the BBB restricts the passage of many therapeutic agents into the CNS. Recent advances in vector design, such as the development of AAV capsids that can effectively cross the BBB, are promising. However, further research is needed to optimize these vectors and ensure safe and efficient delivery of gene therapies to target cells within the CNS [124].
Amyotrophic lateral sclerosis (ALS)
ALS is a chronic neurodegenerative disease with a rapid decline in health conditions and a gradual sudden synapse loss. Patients with ALS characteristically have a loss of motor neurons in the region of the brain, spinal cord, and motor cortex [120, 125]. ALS is the most common muscle disease worldwide. ALS is a neurologic disease that affects motor nerves and progresses over time. Symptoms include muscle weakness, trouble swallowing, and problems with breathing. People with a family history of ALS or FALS are an average of 40 years old, while people without a family history are 50 years old. People 60–69 years of age are most likely to have ALS or FALS [126].
Recabarren-Leiva and Alarcon carefully looked at several genes (DSCR1L1, PCP4, UCHL1, MT1X. GABRA1, EGR1, PCP4, SLC14A1, OLFM1, AQP1, and VSNL1) that are controlled in parts of the spinal cord and motor cortex and are linked to the risk of ALS. The risk of acquiring ALS may be linked to these genes in different ways. These factors are connected with Cytoscape in a network that helps find pathways linked to ALS so that the miRNA and drugs can be identified. The most important route that is changed is PI3K-Akt signaling. As possible parts of the treatment, 13 micro-RNAs have been named (miRNA-328, miRNA-124-1, miRNA-19B2, miRNA-29B2, miRNA-107, miRNA-9-3, miRNA-124-2, miRNA-29A, miRNA-124-3, miRNA-15A, miRNA-9-2, miRNA-19B1, and miRNA-9-1). Four new active drugs have also been named (estradiol, acetaminophen, resveratrol, and progesterone) [127].
The TBK1 gene was analyzed by Tohnai et al., which revealed the occurrence and features of gene variants of TBK1 in Japanese irregular ALS patients, with the help of exome sequencing in 713 sporadic ALS patients and 800 controls. Variants of the TBK1 gene, which are rarely toxic, were found in 9 patients (1.26%) with sporadic ALS, which included four new missense variants, such as p.R358C, p.H322R, p.V23I, and p.T478I, and three other variants with loss-of-function, such as p.P378_I379del, p.R357X, and p.T419_G420del. The average ratio of odds between the sporadic ALS patients and controls was 10.2 (95% CI = 1.67–62.47; P = 0.008). Those outcomes encourage the involvement of TBK1 in Japanese patients in the etiology of sporadic ALS [128].
Nusinersen, an antisense oligonucleotide (ASO), represents a significant advance in the field of gene therapy for neurologic disorders, particularly because nusinersen was the first drug approved by the FDA for the treatment of spinal muscular atrophy (SMA) in both children and adults [129]. This milestone not only highlights the potential of gene therapy to address genetic disorders but also sets a precedent for future therapies targeting similar conditions [130].
SMA is a genetic disorder characterized by the loss of motor neurons in the spinal cord, leading to progressive muscle weakness and atrophy. Nusinersen works by modifying the splicing of the SMN2 gene, which is crucial for the production of the survival motor neuron (SMN) protein. By increasing the levels of functional SMN protein, nusinersen helps slow progression of the disease and improve motor function in patients [129, 131].
The approval of nusinersen has opened new avenues for research and development in gene therapy, demonstrating that targeted genetic interventions can lead to meaningful clinical outcomes. This success story emphasizes the importance of continued investment in gene therapy research because of the promise to transform the treatment landscape for various neurodegenerative disorders [132, 133].
In summary, the role of nusinersen as the first FDA-approved ASO for SMA underscores the potential of gene therapy to provide effective treatments for genetic disorders, paving the way for future innovations in the field.
Future prospects
The field of gene therapy for neurologic disorders is poised for significant advances in the coming years, driven by ongoing research and technological innovations. As we look to the future, several key areas hold promise for enhancing the efficacy and applicability of gene therapy in treating conditions, such as PD, HD, and AD.
- Improved Delivery Mechanisms: One of the primary challenges in gene therapy is the effective delivery of therapeutic agents across the BBB. Future research will likely focus on developing novel delivery systems, including advanced viral vectors and non-viral methods, to enhance the precision and efficiency of gene transfer to target cells in the CNS.
- mRNA Technology: The recent success of mRNA vaccines in combating infectious diseases has opened new avenues for utilizing mRNA technology in gene therapy. This approach could allow for the transient expression of therapeutic proteins, reducing the risk of long-term genomic integration and associated complications. Future studies may explore the application of mRNA-based therapies for neurologic disorders, potentially offering a safer and more flexible treatment option.
- Personalized Medicine: Advances in genomics and biomarker discovery are paving the way for personalized gene therapy approaches. By tailoring treatments based on individual genetic profiles, researchers can enhance the effectiveness of therapies and minimize adverse effects. Future clinical trials may increasingly incorporate personalized strategies to optimize patient outcomes.
- Regulatory and Ethical Considerations: As gene therapy continues to evolve, it will be essential to address the regulatory and ethical implications associated with these innovative treatments. Establishing clear guidelines and frameworks will be crucial for ensuring patient safety and fostering public trust in gene therapy.
- Collaboration and Multidisciplinary Approaches: The complexity of neurologic disorders necessitates collaboration across various fields, including molecular biology, neurology, and bioengineering. Future research initiatives will benefit from interdisciplinary partnerships that leverage diverse expertise to tackle the multifaceted challenges of gene therapy.
The future of gene therapy for neurologic disorders is bright, with numerous opportunities for innovation and improvement. By addressing current challenges and embracing new technologies, the field can move closer to realizing the full potential of gene therapy as a transformative treatment option for patients suffering from these debilitating conditions.
Conclusion
Gene therapy has become an essential and emerging strategy in treating neurodegenerative disorders and is suitable for highly supported genetic targets but not suitable for conventional treatments. In clinical trials for several individual neurodegenerative diseases involving ALS, HD, PD, and AD, this method has been well-tolerated and demonstrated as a long-lasting effect. Additionally, distribution enhancements, like the administration of drugs directly to the CNS, and the carriers are being refined and investigated intensively, such as AAV9 and liposomes. While gene therapy based on non-viral vectors has not been accepted for treating neurodegenerative diseases, recent developments have been generated in clinical trials, which have excited researchers to find new therapies. For the onset and development of neurodegenerative disease, a better understanding method should be promoted for timely detection and selection of targets, allowing early treatment for some of these conditions. The scenarios of gene remedy for neurodegenerative disease would inevitably develop much brighter as development in improving transgene design and vectors. ICV, intrathecal, and direct injection into the spinal cord and brain are some of the delivery ways that are being investigate and made better. These are the patents and clinical studies of different gene therapies that have been studies recently as possible treatments for neurologic diseases (Tables 1 and 2).
Table 2 List of Patents Issued for Several Gene Therapy Applications [133]
| Application Number | Title | Publication Date | Applicants |
|---|---|---|---|
| WO2021076941A1 | Gene therapy for Alzheimer’s disease | 22.04.2021 | Cornell University |
| EP3887396A4 | Gene therapies for neurodegenerative disease | 07.09.2022 | Prevail Therapeutics, Inc. |
| US20230405148A1 | Gene therapy for Alzheimer’s disease | 21.12.2023 | Cornell University |
| WO2023198745A1 | Nucleic acid regulation of APOE | 19.10.2023 | uniQure Biopharma B.V. |
| CA3157864A | Gene therapy for Alzheimer’s disease | 22.04.2022 | Cornell University |
| WO2024011237A1 | Methods and pharmaceutical compositions for the treatment and the prevention of Alzheimer’s disease | 11.01.2024 | Cornell University |
| AU2021200242B2 | AAV vectors for retinal and CNS gene therapy | 21.09.2023 | Genzyme Corp |
| US20230330194A1 | Methods of cytotoxic gene therapy to treat tumors | 19.10.2023 | Candel Therapeutics Inc. |
| EP3193944B1 | Methods of treating cells containing fusion genes | 07.04.2021 | University of Pittsburgh |
| US20230332156A1 | Compositions and methods of treating amyotrophic lateral sclerosis (ALS) | 19.10.2023 | Voyager Therapeutics Inc. |
Declarations
Authors’ contributions
All authors contributed directly and indirectly to the publication of this manuscript.
Financial support
None.
Acknowledgment
All authors thanked Mr. Jitender Joshi (President) and Prof. (Dr.) Dharam Buddhi (Vice Chancellor) of Uttaranchal University for their research-associated encouragement.
Conflicts of interests
No conflicts of interest.
Abbreviations
CNS: Central nervous system; AAVs: Adeno-associated viruses; Adv: Adenovirus; LV: lentivirus, MS: Multiple sclerosis; EAE: Encephalomyelitis; Th1: T-helper type 1; miRNAs: Mammalian non-coding micro-RNAs; nts: Nucleotides; AIDs: Autoimmune diseases; PBMCs: Peripheral blood cells mononuclear cells; HOTAIR: Antisense transcript intervened RNA; HTT: Huntingtin; NPs: Nanoparticles; siRNA: Small interfering RNA; TFs: Transcription factors; GIP: Glucose-dependent insulinotropic polypeptide; m: Mutant; AAV5: Adeno-associated viral serotype 5; AD: Alzheimer’s disease; BBB: Blood-brain barrier; NMDA: N-methyl-D-aspartic acid; APP: Aβ precursor protein; PS1: Presenilin 1; FAD: Family AD; SAD: Sporadic AD; HSV: Herpes simplex virus; shRNA: Short hairpin RNA; PPARγ: PPAR gamma; PGC-1a: PPARγ coactivator one alpha; BDNF: Brain-derived neurotrophic factor; LXRs: Liver X receptors; BRICHOS: Bri2, chondromodulin-I, and SP-C; TDP-43: TAR DNA-binding protein 43; FTD: Frontotemporal dementia; PD: Parkinson’s disease; GABA: Glutamatergic and γ-aminobutyric acid; STN: Subthalamic nucleus; SNpr: Substantia nigra pars reticularis; GPi: Globus pallidus; GCH-1: GTP-cyclohydrolase-1; AADC: Amino acid decarboxylase; TH: Tyrosine hydroxylase; MVs: Modified viruses; NF: Neurotrophic factor; GPe: Globus pallidus; GABA: Gamma-aminobutyric acid; GAD: Glutamate decarboxylase; ALS: Amyotrophic lateral sclerosis.
Graphical abstract
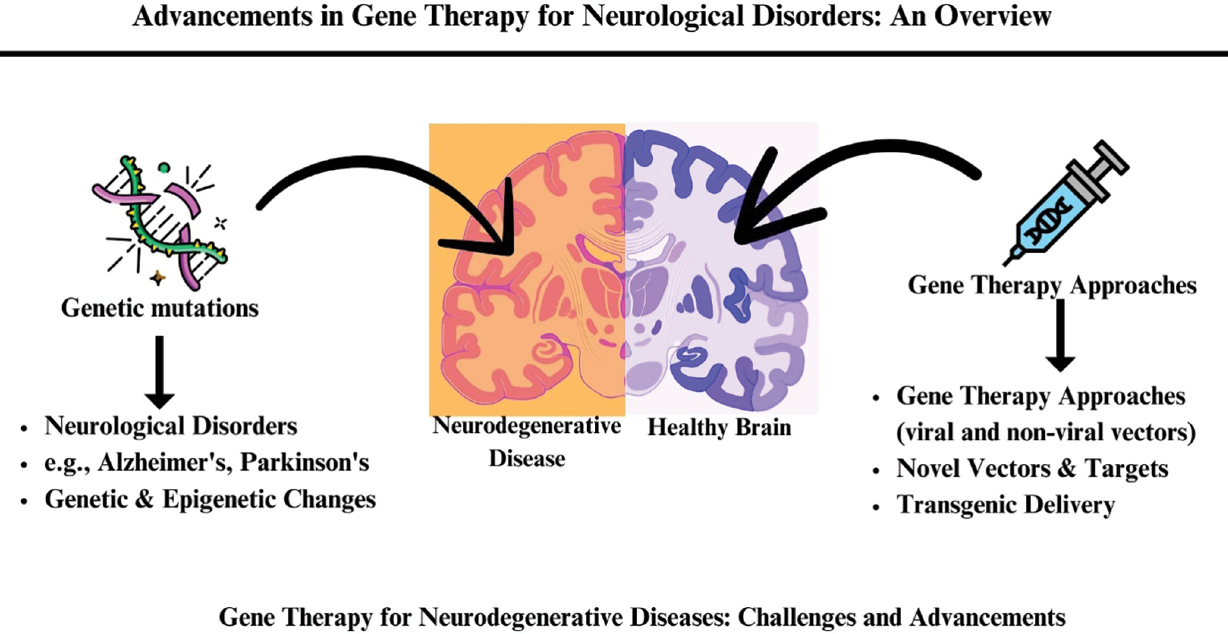
Advances in Gene Therapy for Neurologic Disorders. This abstract illustrates the relationship between genetic mutations leading to neurological disorders such as Alzheimer’s and Parkinson’s diseases and the potential of gene therapy approaches to address these conditions. The left section highlights genetic mutations and their connection to neurodegenerative diseases. The center compares a neurodegenerative disease-affected brain and a healthy brain. The right section outlines various gene therapy approaches, including viral and non-viral vectors, novel vectors and targets, and transgenic delivery methods. The bottom caption emphasizes the challenges and advancements in gene therapy for neurodegenerative diseases.
Highlights
- Over 100 AAV variations, including 13 serotypes (AAV1-13), are used to enhance targeted gene delivery.
- AAV2 is known for its safety and sustained expression in neurons; it has been extensively studied.
- Advanced mechanisms such as ASOs, CRISPR-Cas9, RNAi, and stem cell therapy are being utilized for gene therapy.
- The focus is on neurodegenerative targets, specifically Alzheimer’s, Parkinson’s, and Huntington’s diseases.
- Key challenges include addressing delivery methods, long-term safety, and ethical considerations.
In brief
Gene therapy for neurologic disorders has significantly progressed in developing advanced delivery systems and targeted approaches. Utilizing adeno-associated virus (AAV) serotypes, especially AAV2, has shown promise in achieving sustained gene expression in neurons. Various mechanisms, including viral vectors, antisense oligonucleotides (ASOs), gene editing (CRISPR-Cas9), RNA interference (RNAi), and stem cell therapy, are being explored to address neurodegenerative diseases such as Alzheimer’s, Parkinson’s, and Huntington’s diseases. Despite the advancements, challenges like delivery efficiency, safety, and ethical concerns remain.
References
- Dunbar CE, High KA, Joung JK, Kohn DB, Ozawa K, et al. Gene therapy comes of age. Science 2018;359(6372):eaan4672. [PMID: 29326244 DOI: 10.1126/SCIENCE.AAN4672]
- Hudry E, Vandenberghe LH. Therapeutic AAV gene transfer to the nervous system: a clinical reality. Neuron 2019;102(1):263. [PMID: 30946822 DOI: 10.1016/j.neuron.2019.03.020]
- Wang D, Tai PWL, Gao G. Adeno-associated virus vector as a platform for gene therapy delivery. Nat Rev Drug Discov 2019;18(5):358-78. [PMID: 30710128 DOI: 10.1038/s41573-019-0012-9]
- Lee JH, Wang JH, Chen J, Li F, Edwards TL, et al. Gene therapy for visual loss: opportunities and concerns. Prog Retin Eye Res 2019;68:31-53. [PMID: 30170104 DOI: 10.1016/j.preteyeres.2018.08.003]
- Samaranch L, Salegio EA, San Sebastian W, Kells AP, Bringas JR, et al. Strong cortical and spinal cord transduction after AAV7 and AAV9 delivery into the cerebrospinal fluid of nonhuman primates. Hum Gene Ther 2013;24(5):526-32. [PMID: 23517473 DOI: 10.1089/hum.2013.005]
- Xiang C, Zhang Y, Guo W, Liang XJ. Biomimetic carbon nanotubes for neurological disease therapeutics as inherent medication. Acta Pharm Sin B 2020;10(2):239-48. [PMID: 32082970 DOI: 10.1016/j.apsb.2019.11.003]
- Cearley CN, Vandenberghe LH, Parente MK, Carnish ER, Wilson JM, et al. Expanded repertoire of AAV vector serotypes mediate unique patterns of transduction in mouse brain. Mol Ther 2008;16(10):1710-8. [PMID: 18714307 DOI: 10.1038/mt.2008.166]
- Bartlett JS, Samulski RJ, McCown TJ. Selective and rapid uptake of adeno-associated virus type 2 in brain. Hum Gene Ther 1998;9(8):1181-6. [PMID: 9625257 DOI: 10.1089/hum.1998.9.8-1181]
- Hutson TH, Verhaagen J, Yáñez-Muñoz RJ, Moon LDF. Corticospinal tract transduction: a comparison of seven adeno-associated viral vector serotypes and a non-integrating lentiviral vector. Gene Ther 2012;19(1):49-60. [PMID: 21562590 DOI: 10.1038/gt.2011.71]
- Katz ML, Tecedor L, Chen Y, Williamson BG, Lysenko E, et al. AAV gene transfer delays disease onset in a TPP1-deficient canine model of the late infantile form of Batten disease. Sci Transl Med 2015;7(313):313ra180. [PMID: 26560358 DOI: 10.1126/scitranslmed.aac6191]
- Federici T, Taub JS, Baum GR, Gray SJ, Grieger JC, et al. Robust spinal motor neuron transduction following intrathecal delivery of AAV9 in pigs. Gene Ther 2012;19(8):852-9. [PMID: 21918551 DOI: 10.1038/gt.2011.130]
- Passini MA, Watson DJ, Vite CH, Landsburg DJ, Feigenbaum AL, et al. Intraventricular brain injection of adeno-associated virus type 1 (AAV1) in neonatal mice results in complementary patterns of neuronal transduction to AAV2 and total long-term correction of storage lesions in the brains of beta-glucuronidase-deficient mice. J Virol 2003;77(12):7034-40. [PMID: 12768022 DOI: 10.1128/jvi.77.12.7034-7040.2003]
- Marks WJ, Ostrem JL, Verhagen L, Starr PA, Larson PS, et al. Safety and tolerability of intraputaminal delivery of CERE-120 (adeno-associated virus serotype 2-neurturin) to patients with idiopathic Parkinson’s disease: an open-label, phase I trial. Lancet Neurol 2008;7(5):400-8. [PMID: 18387850 DOI: 10.1016/S1474-4422(08)70065-6]
- Rafii MS, Tuszynski MH, Thomas RG, Barba D, Brewer JB, et al. Adeno-associated viral vector (serotype 2)-nerve growth factor for patients with Alzheimer disease: a randomized clinical trial. JAMA Neurol 2018;75(7):834-41. [PMID: 29582053 DOI: 10.1001/jamaneurol.2018.0233]
- Adachi K, Enoki T, Kawano Y, Veraz M, Nakai H. Drawing a high-resolution functional map of adeno-associated virus capsid by massively parallel sequencing. Nat Commun 2014;5:3075. [PMID: 24435020 DOI: 10.1038/ncomms4075]
- Albright BH, Storey CM, Murlidharan G, Castellanos Rivera RM, Berry GE, et al. Mapping the structural determinants required for AAVrh.10 transport across the blood-brain barrier. Mol Ther 2018;26(2):510-23. [PMID: 29175157 DOI: 10.1016/j.ymthe.2017.10.017]
- Allen WE, Kauvar IV, Chen MZ, Richman EB, Yang SJ, et al. Global representations of goal-directed behavior in distinct cell types of mouse neocortex. Neuron 2017;94(4):891-907.e6. [PMID: 28521139 DOI: 10.1016/j.neuron.2017.04.017]
- Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, et al. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med 2013;5(189):189ra76. [PMID: 23761039 DOI: 10.1126/scitranslmed.3005708]
- Barkats M, Bilang-Bleuel A, Buc-Caron MH, Castel-Barthe MN, Corti O, et al. Adenovirus in the brain: recent advances of gene therapy for neurodegenerative diseases. Prog Neurobiol 1998;55(4):333-41. [PMID: 9654383 DOI: 10.1016/s0301-0082(98)00028-8]
- Kritzinger A, Ferger B, Gillardon F, Stierstorfer B, Birk G, et al. Age-related pathology after adenoviral overexpression of the leucine-rich repeat kinase 2 in the mouse striatum. Neurobiol Aging 2018;66:97-111. [PMID: 29550548 DOI: 10.1016/j.neurobiolaging.2018.02.008]
- Pack DW, Hoffman AS, Pun S, Stayton PS. Design and development of polymers for gene delivery. Nat Rev Drug Discov 2005;4(7):581-93. [PMID: 16052241 DOI: 10.1038/nrd1775]
- Mintzer MA, Simanek EE. Nonviral vectors for gene delivery. Chem Rev 2009;109(2):259-302. [PMID: 19053809 DOI: 10.1021/cr800409e]
- Buck J, Grossen P, Cullis PR, Huwyler J, Witzigmann D. Lipid-based DNA therapeutics: hallmarks of non-viral gene delivery. ACS Nano 2019;13(4):3754-82. [PMID: 30908008 DOI: 10.1021/acsnano.8b07858]
- Yin H, Kanasty RL, Eltoukhy AA, Vegas AJ, Dorkin JR, et al. Non-viral vectors for gene-based therapy. Nat Rev Genet 2014;15(8):541-55. [PMID: 25022906 DOI: 10.1038/nrg3763]
- Li W, Szoka FC. Lipid-based nanoparticles for nucleic acid delivery. Pharm Res 2007;24(3):438-49. [PMID: 17252188 DOI: 10.1007/s11095-006-9180-5]
- Conceição M, Mendonça L, Nóbrega C, Gomes C, Costa P, et al. Intravenous administration of brain-targeted stable nucleic acid lipid particles alleviates Machado-Joseph disease neurological phenotype. Biomaterials 2016;82:124-37. [PMID: 26757259 DOI: 10.1016/j.biomaterials.2015.12.021]
- Carradori D, Eyer J, Saulnier P, Préat V, des Rieux A. The therapeutic contribution of nanomedicine to treat neurodegenerative diseases via neural stem cell differentiation. Biomaterials 2017;123:77-91. [PMID: 28161683 DOI: 10.1016/j.biomaterials.2017.01.032]
- Niu S, Zhang LK, Zhang L, Zhuang S, Zhan X, et al. Inhibition by multifunctional magnetic nanoparticles loaded with alpha-synuclein RNAi plasmid in a Parkinson’s disease model. Theranostics 2017;7(2):344-56. [PMID: 28042339 DOI: 10.7150/thno.16562]
- Semple SC, Akin A, Chen J, Sandhu AP, Mui BL, et al. Rational design of cationic lipids for siRNA delivery. Nat Biotechnol 2010;28(2):172-6. [PMID: 20081866 DOI: 10.1038/nbt.1602]
- Kojima R, Bojar D, Rizzi G, Hamri GC, El-Baba MD, et al. Designer exosomes produced by implanted cells intracerebrally deliver therapeutic cargo for Parkinson’s disease treatment. Nat Commun 2018;9(1):1305. [PMID: 29610454 DOI: 10.1038/s41467-018-03733-8]
- Chen W, Luan J, Wei G, Zhang X, Fan J, et al. In vivo hepatocellular expression of interleukin-22 using penetratin-based hybrid nanoparticles as potential anti-hepatitis therapeutics. Biomaterials 2018;187:66-80. [PMID: 30296739 DOI: 10.1016/j.biomaterials.2018.09.046]
- Morris VB, Labhasetwar V. Arginine-rich polyplexes for gene delivery to neuronal cells. Biomaterials 2015;60:151-60. [PMID: 26000961 DOI: 10.1016/j.biomaterials.2015.04.052]
- Malhotra M, Tomaro-Duchesneau C, Prakash S. Synthesis of TAT peptide-tagged PEGylated chitosan nanoparticles for siRNA delivery targeting neurodegenerative diseases. Biomaterials 2013;34(4):1270-80. [PMID: 23140978 DOI: 10.1016/j.biomaterials.2012.10.013]
- Hitti FL, Gonzalez-Alegre P, Lucas TH. Gene therapy for neurologic disease: a neurosurgical review. World Neurosurg 2019;121:261-73. [PMID: 30253990 DOI: 10.1016/j.wneu.2018.09.097]
- Cree BAC. Multiple sclerosis genetics. Mult Scler Relat Disord 2018. [DOI: 10.1891/9780826125941.0005]
- Correale J, Gaitán MI, Ysrraelit MC, Fiol MP. Progressive multiple sclerosis: from pathogenic mechanisms to treatment. Brain 2017;140(3):527-46. [PMID: 27794524 DOI: 10.1093/brain/aww258]
- Capone A, Bianco M, Ruocco G, de Bardi M, Battistini L, et al. Distinct expression of inflammatory features in T helper 17 cells from multiple sclerosis patients. Cells 2019;8:533. [PMID: 31167379 DOI: 10.3390/cells8060533]
- Li QW, Lei W, Chen C, Guo W. Recent advances of long noncoding RNAs involved in the development of multiple sclerosis. Chin J Nat Med 2020;18(1):36-46. [PMID: 31955822 DOI: 10.1016/S1875-5364(20)30003-0]
- Piket E, Zheleznyakova GY, Kular L, Jagodic M. Small non-coding RNAs as important players, biomarkers and therapeutic targets in multiple sclerosis: a comprehensive overview. J Autoimmun 2019;101:17-25. [PMID: 31014917 DOI: 10.1016/j.jaut.2019.04.002]
- Fenoglio C, Cantoni C, De Riz M, Ridolfi E, Cortini F, et al. Expression and genetic analysis of miRNAs involved in CD4+ cell activation in patients with multiple sclerosis. Neurosci Lett 2011;504(1):9-12. [PMID: 21875645 DOI: 10.1016/j.neulet.2011.08.021]
- Rupaimoole R, Slack FJ. MicroRNA therapeutics: towards a new era for the management of cancer and other diseases. Nat Rev Drug Discov 2017;16(3):203-22. [PMID: 28209991 DOI: 10.1038/nrd.2016.246]
- Mohamed MS, Nahrery EMAE, Shalaby N, Hussein M, Aal RAE, et al. Micro-RNA 18b and interleukin 17A profiles in relapsing remitting multiple sclerosis. Mult Scler Relat Disord 2019;28:226-9. [PMID: 30623862 DOI: 10.1016/j.msard.2018.12.013]
- Zhang F, Liu G, Wei C, Gao C, Hao J. Linc-MAF-4 regulates Th1/Th2 differentiation and is associated with the pathogenesis of multiple sclerosis by targeting MAF. FASEB J 2017;31(2):519-25. [PMID: 27756768 DOI: 10.1096/fj.201600838R]
- Ranzani V, Rossetti G, Panzeri I, Arrigoni A, Bonnal RJ, et al. The long intergenic noncoding RNA landscape of human lymphocytes highlights the regulation of T cell differentiation by linc-MAF-4. Nat Immunol 2015;16(3):318-25. [PMID: 25621826 DOI: 10.1038/ni.3093]
- Portoso M, Ragazzini R, Brenčič Ž, Moiani A, Michaud A, et al. PRC2 is dispensable for HOTAIR-mediated transcriptional repression. EMBO J 2017;36(8):981-94. [PMID: 28167697 DOI: 10.15252/embj.201695335]
- Song J, Kim D, Han J, Kim Y, Lee M, et al. PBMC and exosome-derived Hotair is a critical regulator and potent marker for rheumatoid arthritis. Clin Exp Med 2015;15:121-6. [PMID: 24722995 DOI: 10.1007/s10238-013-0271-4]
- Li Z, Li X, Jiang C, Qian W, Tse G, et al. Long non-coding RNAs in rheumatoid arthritis. Cell Prolif 2018;51:e12404. [PMID: 29110355 DOI: 10.1111/cpr.12404]
- Pahlevan Kakhki M, Nikravesh A, Shirvani Farsani Z, Sahraian MA, Behmanesh M. HOTAIR but not ANRIL long non-coding RNA contributes to the pathogenesis of multiple sclerosis. Immunology 2018;153:479-87. [PMID: 29030863 DOI: 10.1111/imm.12850]
- Sun S, Wu Y, Guo W, Yu F, Kong L, et al. STAT3/HOTAIR signaling axis regulates HNSCC growth in an EZH2-dependent manner. Clin Cancer Res 2018;24:2665-77. [PMID: 29540490 DOI: 10.1158/1078-0432.CCR-16-2248]
- Guo W, Luo C, Wang C, Zhu Y, Wang X, et al. Protection against Th17 cells differentiation by an interleukin-23 receptor cytokine-binding homology region. PLoS One 2012;7:e45625. [PMID: 23029144 DOI: 10.1371/journal.pone.0045625]
- Guo W, Luo C, Wang C, Wang YH, Wang X, et al. Suppression of human and mouse Th17 differentiation and autoimmunity by an endogenous Interleukin 23 receptor cytokine-binding homology region. Int J Biochem Cell Biol 2014;55:304-10. [PMID: 25263529 DOI: 10.1016/j.biocel.2014.09.019]
- Guo W, Lei W, Yu D, Ge Y, Chen Y, et al. Involvement of lncRNA-1700040D17Rik in Th17 cell differentiation and the pathogenesis of EAE. Int Immunopharmacol 2017;47:141-9. [PMID: 28395256 DOI: 10.1016/j.intimp.2017.03.014]
- Aylward EH, Codori AM, Rosenblatt A, Sherr M, Brandt J, et al. Rate of caudate atrophy in presymptomatic and symptomatic stages of Huntington’s disease. Mov Disord 2000;15:552-60. [PMID: 10830423 DOI: 10.1002/1531-8257(200005)15:3[[552::AID-MDS1020]]3.0.CO;2-P]
- MacDonald ME, Ambrose CM, Duyao MP, Myers RH, Lin C, et al. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell 1993;72:971-83. [DOI: 10.1016/0092-8674(93)90585-E]
- Rawlins MD, Wexler NS, Wexler AR, Tabrizi SJ, Douglas I, et al. The prevalence of Huntington’s disease. Neuroepidemiology 2016;46:144-53. [PMID: 26824438 DOI: 10.1159/000443738]
- Roos RAC. Huntington’s disease: a clinical review. Orphanet J Rare Dis 2010;5:40. [PMID: 21171977 DOI: 10.1186/1750-1172-5-40]
- Landles C, Bates GP. Huntingtin and the molecular pathogenesis of Huntington’s disease. EMBO Rep 2004;5:958-63. [PMID: 15459747 DOI: 10.1038/sj.embor.7400250]
- Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S, et al. Quantification of mutant huntingtin protein in cerebrospinal fluid from Huntington’s disease patients. J Clin Invest 2015;125:1979-86. [PMID: 25844897 DOI: 10.1172/JCI80743]
- Sava V, Fihurka O, Khvorova A, Sanchez-Ramos J. Enriched chitosan nanoparticles loaded with siRNA are effective in lowering Huntington’s disease gene expression following intranasal administration. Nanomedicine 2020;24:102119. [PMID: 31666200 DOI: 10.1016/j.nano.2019.102119]
- Kacher R, Lamazière A, Heck N, Kappes V, Mounier C, et al. CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington’s disease. Brain 2019;142:2432-50. [PMID: 31286142 DOI: 10.1093/brain/awz174]
- Spronck EA, Brouwers CC, Vallès A, de Haan M, Petry H, et al. AAV5-miHTT gene therapy demonstrates sustained Huntingtin lowering and functional improvement in Huntington disease mouse models. Mol Ther Methods Clin Dev 2019;13:334-43. [PMID: 30984798 DOI: 10.1016/j.omtm.2019.03.002]
- Zeitler B, Froelich S, Marlen K, Shivak DA, Yu Q, et al. Allele-selective transcriptional repression of mutant HTT for the treatment of Huntington’s disease. Nat Med 2019;25:1131-42. [PMID: 31263285 DOI: 10.1038/s41591-019-0478-3]
- Verma MK, Goel R, Nandakumar K, Nemmani KVS. Bilateral quinolinic acid-induced lipid peroxidation, decreased striatal monoamine levels and neurobehavioral deficits are ameliorated by GIP receptor agonist D-Ala2GIP in rat model of Huntington’s disease. Eur J Pharmacol 2018;828:31-41. [PMID: 29577894 DOI: 10.1016/j.ejphar.2018.03.034]
- Pfister EL, Dinardo N, Mondo E, Borel F, Conroy F, et al. Artificial miRNAs reduce human mutant Huntingtin throughout the striatum in a transgenic sheep model of Huntington’s disease. Hum Gene Ther 2018;29:663-73. [PMID: 29207890 DOI: 10.1089/hum.2017.199]
- Yang S, Li S, Li X, Yang S, Chang R, et al. CRISPR/Cas9-mediated gene editing ameliorates neurotoxicity in mouse model of Huntington’s disease. J Clin Invest 2017;127:2719-24. [PMID: 28628038 DOI: 10.1172/JCI92087]
- Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol 2010;22:124-31. [PMID: 20034776 DOI: 10.1016/j.ceb.2009.11.014]
- Freude K, Krauss S. Dementia, brain disorders and molecular mechanisms. J Mol Biol 2019;431:1709-10. [PMID: 30930050 DOI: 10.1016/j.jmb.2019.03.025]
- Winblad B, Amouyel P, Andrieu S, Ballard C, Brayne C, et al. Defeating Alzheimer’s disease and other dementias: a priority for European science and society. Lancet Neurol 2016;15:455-532. [PMID: 26987701 DOI: 10.1016/S1474-4422(16)00062-4]
- Rodriguez-Vieitez E, Saint-Aubert L, Carter SF, Almkvist O, Farid K, et al. Diverging longitudinal changes in astrocytosis and amyloid PET in autosomal dominant Alzheimer’s disease. Brain 2016;139:922-36. [PMID: 26813969 DOI: 10.1093/brain/awv404]
- Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. J Cell Sci 2007;120:4081-91. [PMID: 18032783 DOI: 10.1242/jcs.019265]
- Hong CS, Goins WF, Goss JR, Burton EA, Glorioso JC. Herpes simplex virus RNAi and neprilysin gene transfer vectors reduce accumulation of Alzheimer’s disease-related amyloid-β peptide in vivo. Gene Ther 2006;13:1068-79. [PMID: 16541122 DOI: 10.1038/sj.gt.3302719]
- Senechal Y, Kelly PH, Cryan JF, Natt F, Dev KK. Amyloid precursor protein knockdown by siRNA impairs spontaneous alternation in adult mice. J Neurochem 2007;102:1928-40. [PMID: 17540010 DOI: 10.1111/j.1471-4159.2007.04672.x]
- Alvarez-Erviti L, Seow Y, Yin H, Betts C, Lakhal S, et al. Delivery of siRNA to the mouse brain by systemic injection of targeted exosomes. Nat Biotechnol 2011;29:341-5. [PMID: 21423189 DOI: 10.1038/nbt.1807]
- Singer O, Marr RA, Rockenstein E, Crews L, Coufal NG, et al. Targeting BACE1 with siRNAs ameliorates Alzheimer disease neuropathology in a transgenic model. Nat Neurosci 2005;8:1343-9. [PMID: 16136043 DOI: 10.1038/nn1531]
- Hu X, Zhou X, He W, Yang J, Xiong W, et al. BACE1 deficiency causes altered neuronal activity and neurodegeneration. J Neurosci 2010;30:8819-29. [PMID: 20592204 DOI: 10.1523/JNEUROSCI.1334-10.2010]
- Fitz NF, Cronican AA, Saleem M, Fauq AH, Chapman R, et al. Abca1 deficiency affects Alzheimer’s disease-like phenotype in human ApoE4 but not in ApoE3-targeted replacement mice. J Neurosci 2012;32:13125-36. [PMID: 22993429 DOI: 10.1523/JNEUROSCI.1937-12.2012]
- Donkin JJ, Stukas S, Hirsch-Reinshagen V, Namjoshi D, Wilkinson A, et al. ATP-binding cassette transporter A1 mediates the beneficial effects of the liver X receptor agonist GW3965 on object recognition memory and amyloid burden in amyloid precursor protein/presenilin 1 mice. J Biol Chem 2010;285:34144-54. [PMID: 20739291 DOI: 10.1074/jbc.M110.108100]
- Zelcer N, Khanlou N, Clare R, Jiang Q, Reed-Geaghan EG, et al. Attenuation of neuroinflammation and Alzheimer’s disease pathology by liver x receptors. Proc Natl Acad Sci U S A 2007;104:10601-6. [PMID: 17563384 DOI: 10.1073/pnas.0701096104]
- Egan CM, Nyman U, Skotte J, Streubel G, Turner S, et al. CHD5 is required for neurogenesis and has a dual role in facilitating gene expression and polycomb gene repression. Dev Cell 2013;26:223-36. [PMID: 23948251 DOI: 10.1016/j.devcel.2013.07.008]
- Sánchez-Pulido L, Devos D, Valencia A. BRICHOS: a conserved domain in proteins associated with dementia, respiratory distress and cancer. Trends Biochem Sci 2002;27:329-32. [PMID: 12114016 DOI: 10.1016/s0968-0004(02)02134-5]
- Willander H, Askarieh G, Landreh M, Westermark P, Nordling K, et al. High-resolution structure of a BRICHOS domain and its implications for anti-amyloid chaperone activity on lung surfactant protein C. Proc Natl Acad Sci U S A 2012;109:2325-9. [PMID: 22308375 DOI: 10.1073/pnas.1114740109]
- Willander H, Hermansson E, Johansson J, Presto J. BRICHOS domain associated with lung fibrosis, dementia and cancer – a chaperone that prevents amyloid fibril formation? FEBS J 2011;278:3893-904. [PMID: 21668643 DOI: 10.1111/j.1742-4658.2011.08209.x]
- Hermansson E, Schultz S, Crowther D, Linse S, Winblad B, et al. The chaperone domain BRICHOS prevents CNS toxicity of amyloid-β peptide in Drosophila melanogaster. Dis Model Mech 2014;7:659-65. [PMID: 24682783 DOI: 10.1242/dmm.014787]
- Arosio P, Michaels T, Linse S, Månsson C, Emanuelsson C, et al. Kinetic analysis reveals the diversity of microscopic mechanisms through which molecular chaperones suppress amyloid formation. Nat Commun 2016;7:10948. [PMID: 27009901 DOI: 10.1038/ncomms10948]
- Kim J, Chakrabarty P, Hanna A, March A, Dickson DW, et al. Normal cognition in transgenic BRI2-Aβ mice. Mol Neurodegener 2013;8:15. [PMID: 23663320 DOI: 10.1186/1750-1326-8-15]
- Cohen SIA, Arosio P, Presto J, Kurudenkandy FR, Biverstål H, et al. A molecular chaperone breaks the catalytic cycle that generates toxic Aβ oligomers. Nat Struct Mol Biol 2015;22:207-13. [PMID: 25686087 DOI: 10.1038/nsmb.2971]
- Poska H, Haslbeck M, Kurudenkandy FR, Hermansson E, Chen G, et al. Dementia-related Bri2 BRICHOS is a versatile molecular chaperone that efficiently inhibits Aβ42 toxicity in Drosophila. Biochem J 2016;473:3683-704. [PMID: 27514716 DOI: 10.1042/BCJ20160277]
- Menzies FM, Fleming A, Rubinsztein DC. Compromised autophagy and neurodegenerative diseases. Nat Rev Neurosci 2015;16:345-57. [PMID: 25991442 DOI: 10.1038/nrn3961]
- Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, et al. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 2007;282:24131-45. [PMID: 17580304 DOI: 10.1074/jbc.M702824200]
- Komatsu M, Waguri S, Chiba T, Murata S, Iwata JI, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 2006;441:880-4. [PMID: 16625205 DOI: 10.1038/nature04723]
- Kim M, Sandford E, Gatica D, Qiu Y, Liu X, et al. Mutation in ATG5 reduces autophagy and leads to ataxia with developmental delay. Elife 2016;5:e12245. [PMID: 26812546 DOI: 10.7554/elife.12245]
- Nakamura Y, Saigoh K, Sakamoto H, Ueno S, Isono C, et al. Mutations in the gene encoding p62 in Japanese patients with amyotrophic lateral sclerosis. Neurology 2013;80:458-63. [PMID: 23303844 DOI: 10.1212/WNL.0b013e31827f0fe5]
- le Ber I, Camuzat A, Guerreiro R, Bouya-Ahmed K, Bras J, et al. SQSTM1 Mutations in French patients with frontotemporal dementia or frontotemporal dementia with amyotrophic lateral sclerosis. JAMA Neurol 2013;70:1403-10. [PMID: 24042580 DOI: 10.1001/jamaneurol.2013.3849]
- Nilsson P, Loganathan K, Sekiguchi M, Matsuba Y, Hui K, et al. Aβ secretion and plaque formation depend on autophagy. Cell Rep 2013;5:61-9. [PMID: 24095740 DOI: 10.1016/j.celrep.2013.08.042]
- Wichmann T, DeLong MR. Pathophysiology of Parkinson’s disease: the MPTP primate model of the human disorder. Ann N Y Acad Sci 2003;991:199-213. [PMID: 12846988 DOI: 10.1111/j.1749-6632.2003.tb07477.x]
- Alvarez L, Macias R, Guridi J, Lopez G, Alvarez E, et al. Dorsal subthalamotomy for Parkinson’s disease. Mov Disord 2001;16:72-8. [PMID: 11215596 DOI: 10.1002/1531-8257(200101)16:1[[72::aid-mds1019]]3.0.co;2-6]
- Su PC, Tseng HM, Liu HM, Yen RF, Liou HH. Treatment of advanced Parkinson’s disease by subthalamotomy: one-year results. Mov Disord 2003;18:531-8. [PMID: 12722167 DOI: 10.1002/mds.10393]
- Obeso JA. Deep-brain stimulation of the subthalamic nucleus or the pars interna of the globus pallidus in Parkinson’s disease. N Engl J Med 2019;345:956-63. [PMID: 11575287 DOI: 10.1056/NEJMoa000827]
- Levy R, Lang AE, Dostrovsky JO, Pahapill P, Romas J, et al. Lidocaine and muscimol microinjections in subthalamic nucleus reverse Parkinsonian symptoms. Brain 2001;124:2105-18. [PMID: 11571226 DOI: 10.1093/brain/124.10.2105]
- Jarraya B, Boulet S, Ralph GS, Jan C, Bonvento G, et al. Dopamine gene therapy for Parkinson’s disease in a nonhuman primate without associated dyskinesia. Sci Transl Med 2009;1:2ra4. [PMID: 20368163 DOI: 10.1126/scitranslmed.3000130]
- Warrington KH, Herzog RW. Treatment of human disease by adeno-associated viral gene transfer. Hum Genet 2006;119:571-603. [PMID: 16612615 DOI: 10.1007/s00439-006-0165-6]
- Sengupta R, Mukherjee C, Sarkar N, Sun Z, Lesnik J, et al. An optimized protocol for packaging pseudotyped Integrase defective lentivirus. Biol Proced Online 2016;18:14. [PMID: 27403084 DOI: 10.1186/s12575-016-0044-z]
- Latchman DS. Gene delivery and gene therapy with herpes simplex virus-based vectors. Gene 2001;264:1-9. [PMID: 11245972 DOI: 10.1016/s0378-1119(01)00322-5]
- Hocquemiller M, Giersch L, Audrain M, Parker S, Cartier N. Adeno-associated virus-based gene therapy for CNS diseases. Hum Gene Ther 2016;27:478-96. [PMID: 27267688 DOI: 10.1089/hum.2016.087]
- Marquez Loza LI, Yuen EC, McCray PB. Lentiviral vectors for the treatment and prevention of cystic fibrosis lung disease. Genes (Basel) 2019;10:218. [PMID: 30875857 DOI: 10.3390/genes10030218]
- Sehara Y, Fujimoto KI, Ikeguchi K, Katakai Y, Ono F, et al. Persistent expression of dopamine-synthesizing enzymes 15 years after gene transfer in a primate model of Parkinson’s disease. Hum Gene Ther Clin Dev 2017;28:74-9. [PMID: 28279081 DOI: 10.1089/humc.2017.010]
- Nagatsua T, Sawadab M. L-Dopa therapy for Parkinson’s disease: past, present, and future. Parkinsonism Relat Disord 2009;15:S3-8. [PMID: 19131039 DOI: 10.1016/S1353-8020(09)70004-5]
- Christine CW, Starr PA, Larson PS, Eberling JL, Jagust WJ, et al. Safety and tolerability of putaminal AADC gene therapy for Parkinson disease. Neurology 2009;73:1662-9. [PMID: 19828868 DOI: 10.1212/WNL.0b013e3181c29356]
- Eberling JL, Jagust WJ, Christine CW, Starr P, Larson P, et al. Results from a phase I safety trial of hAADC gene therapy for Parkinson disease. Neurology 2008;70:1980-3. [PMID: 18401019 DOI: 10.1212/01.wnl.0000312381.29287.ff]
- Muramatsu SI, Fujimoto KI, Kato S, Mizukami H, Asari S, et al. A phase I study of aromatic L-amino acid decarboxylase gene therapy for Parkinson’s disease. Mol Therapy 2010;18:1731-5. [PMID: 20606642 DOI: 10.1038/mt.2010.135]
- Christine CW, Bankiewicz KS, Van Laar AD, Richardson RM, Ravina B, et al. Magnetic resonance imaging–guided phase 1 trial of putaminal AADC gene therapy for Parkinson’s disease. Ann Neurol 2019;85:704-14. [PMID: 30802998 DOI: 10.1002/ana.25450]
- Han SJ, Bankiewicz K, Butowski NA, Larson PS, Aghi MK. Interventional MRI-guided catheter placement and real time drug delivery to the central nervous system. Expert Rev Neurother 2016;16:635-9. [PMID: 27054877 DOI: 10.1080/14737175.2016.1175939]
- Gill SS, Patel NK, Hotton GR, O’Sullivan K, McCarter R, et al. Direct brain infusion of glial cell line-derived neurotrophic factor in Parkinson disease. Nat Med 2003;9:589-95. [PMID: 12669033 DOI: 10.1038/nm850]
- Whone A, Luz M, Boca M, Woolley M, Mooney L, et al. Randomized trial of intermittent intraputamenal glial cell line-derived neurotrophic factor in Parkinson’s disease. Brain 2019;142:512-25. [PMID: 30808022 DOI: 10.1093/brain/awz023]
- Whone AL, Boca M, Luz M, Woolley M, Mooney L, et al. Extended treatment with glial cell line-derived neurotrophic factor in Parkinson’s disease. J Parkinsons Dis 2019;9:301-13. [PMID: 30829619 DOI: 10.3233/JPD-191576]
- Review N, Circuits BG. Circuits and circuit disorders of the basal ganglia. Arch Neurol 2007;64:20-4. [PMID: 17210805 DOI: 10.1001/archneur.64.1.20]
- Obeso JA, Rodriguez-Oroz MC, Rodriguez M, Macias R, Alvarez L, et al. Pathophysiologic basis of surgery for Parkinson’s disease. Neurology 2000;55:S7-12. [PMID: 11188978]
- Erlander MG, Tillakaratne NJK, Feldblum S, Patel N, Tobin AJ. Two genes encode distinct glutamate decarboxylases. Neuron 1991;7:91-100. [PMID: 2069816 DOI: 10.1016/0896-6273(91)90077-d]
- Münster-Wandowski A, Zander J-F, Richter K, Ahnert-Hilger G. Co-existence of functionally different vesicular neurotransmitter transporters. Front Synaptic Neurosci 2016;8:4. [PMID: 26909036 DOI: 10.3389/fnsyn.2016.00004]
- Roccaro-Waldmeyer DM, Girard F, Milani D, Vannoni E, Prétôt L, et al. Eliminating the VGlut2-dependent glutamatergic transmission of parvalbumin-expressing neurons leads to deficits in locomotion and vocalization, decreased pain sensitivity, and increased dominance. Front Behav Neurosci 2018;12:146. [PMID: 30072881 DOI: 10.3389/fnbeh.2018.00146]
- Wang JH, Gessler DJ, Zhan W, Gallagher TL, Gao G. Adeno-associated virus as a delivery vector for gene therapy of human diseases. Sig Transduct Target Ther 2024;9:1-33. [DOI: 10.1038/s41392-024-01780-w]
- Wu D, Chen Q, Chen X, Han F, Chen Z, et al. The blood–brain barrier: structure, regulation and drug delivery. Sig Transduct Target Ther 2023;8:217. [PMID: 37231000 DOI: 10.1038/s41392-023-01481-w]
- Ndemazie NB, Inkoom A, Morfaw EF, Smith T, Aghimien M, et al. Multi-disciplinary approach for drug and gene delivery systems to the brain. AAPS PharmSciTech 2021;23:11. [PMID: 34862567 DOI: 10.1208/s12249-021-02144-1]
- Gao Z. Strategies for enhanced gene delivery to the central nervous system. Nanoscale Adv 2024;6:3009-28. [PMID: 38868835 DOI: 10.1039/d3na01125a]
- Riva N, Agosta F, Lunetta C, Filippi M, Quattrini A. Recent advances in amyotrophic lateral sclerosis. J Neurol 2016;263:1241-54. [PMID: 27025851 DOI: 10.1007/s00415-016-8091-6]
- Vucic S, Rothstein JD, Kiernan MC. Advances in treating amyotrophic lateral sclerosis: insights from pathophysiological studies. Trends Neurosci 2014;37:433-42. [PMID: 24927875 DOI: 10.1016/j.tins.2014.05.006]
- Recabarren-Leiva D, Alarcón M. New insights into the gene expression associated to amyotrophic lateral sclerosis. Life Sci 2018;193:110-23. [PMID: 29241710 DOI: 10.1016/j.lfs.2017.12.016]
- Tohnai G, Nakamura R, Sone J, Nakatochi M, Yokoi D, et al. Frequency and characteristics of the TBK1 gene variants in Japanese patients with sporadic amyotrophic lateral sclerosis. Neurobiol Aging 2018;64:158.e15-9. [PMID: 29398122 DOI: 10.1016/j.neurobiolaging.2017.12.005]
- Nakevska Z, Yokota T. Challenges and future perspective of antisense therapy for spinal muscular atrophy: a review. Eur J Cell Biol 2023;102:151326. [PMID: 37295266 DOI: 10.1016/j.ejcb.2023.151326]
- Mendell JR, Al-Zaidy S, Shell R, Arnold WD, Rodino-Klapac LR, et al. Single-dose gene-replacement therapy for spinal muscular atrophy. N Engl J Med 2017;377:1713-22. [PMID: 29091557 DOI: 10.1056/NEJMoa1706198]
- Aslesh T, Yokota T. Restoring SMN expression: an overview of the therapeutic developments for the treatment of spinal muscular atrophy. Cells 2022;11:417. [PMID: 35159227 DOI: 10.3390/cells11030417]
- Qiu J, Wu L, Qu R, Jiang T, Bai J, et al. History of development of the life-saving drug “Nusinersen” in spinal muscular atrophy. Front Cell Neurosci 2022;16:942976. [PMID: 36035257 DOI: 10.3389/fncel.2022.942976]
- Haque US, Yokota T. Recent progress in gene-targeting therapies for spinal muscular atrophy: promises and challenges. Genes 2024;15:999. [PMID: 39202360 DOI: 10.3390/genes15080999]