Integration Analysis of m6A Regulators and m6A-Related Genes in Hepatocellular Carcinoma
1Department of Anesthesiology, Sun Yat-sen University Cancer Center, State Key Laboratory of Oncology in Southern China, Collaborative Innovation for Cancer Medicine, Guangzhou, Guangdong 510000, China
aThese authors contributed equally to this work.
*Correspondence: Qiang Li, E-mail: liqiang@sysucc.org.cn; Dongtai Chen, E-mail: chendt@sysucc.org.cn
Received: January 13 2021; Revised: January 30 2021; Accepted: February 10 2021; Published Online: March 3 2021
Cite this paper:
Jingdun Xie, Zhenhua Qi, Xiaolin Luo, Fang Yan, Wei Xing, Weian Zeng, Dongtai Chen and Qiang Li. Integration Analysis of m6A Regulators and m6A-Related Genes in Hepatocellular Carcinoma. BIO Integration 2021; 2(3): 94–108.
DOI: 10.15212/bioi-2021-0002. Available at: https://bio-integration.org/
Download citation 
© 2021 The Authors. This is an open access article distributed under the terms of the Creative Commons Attribution License (https://creativecommons.org/licenses/by/4.0/). See https://bio-integration.org/copyright-and-permissions/
Abstract
Background: N6-Methyladenosine (m6A) RNA methylation of eukaryotic mRNA is involved in the progression of various tumors. We aimed to investigate m6A-related genes and m6A regulators in hepatocellular carcinoma (HCC) and their association with prognosis in HCC.
Methods: We downloaded liver cancer sample data from The Cancer Genome Atlas (TCGA) and the International Cancer Genome Consortium database. A total of 21 m6A regulators and 1258 m6A-related genes were then analyzed by consensus clustering, Spearman’s correlation, GO, KEGG, LASSO Cox regression, and univariate Cox regression analyses. Finally, we constructed a risk prognostic model.
Results: We obtained 192 candidate m6A-related genes and 3 m6A regulators, including YTHDF1, YTHDF2, and YTHDC1. The expression of these genes and regulators differed significantly in different stages of HCC. Based on Cox regression analysis, 19 of 98 m6A-related prognostic genes were obtained to construct a risk score model. The 1- and 3-year area under the curves (AUCs) among HCC patients were greater than 0.7. Finally, based on analysis of mutation differences between high- and low-risk score groups, we determined that TP53 had the highest mutation frequency in the high-risk HCC patient group, whereas titin (TTN) had the highest mutation frequency in the low-risk HCC patient group.
Conclusion: This study comprehensively analyzed m6A regulators and m6A-related genes through an integrated bioinformatic analysis, including expression, clustering, protein–protein interaction, and prognosis, thus providing novel insights into the roles of m6A regulators and m6A-related genes in HCC.
Keywords
Hepatocellular carcinoma, m6A-related genes, m6A regulators, prognosis.
Introduction
Hepatocellular carcinoma (HCC), which ranks fourth in deaths caused by cancer [1], causes over 600,000 deaths annually worldwide [2]. Over the past few years, considerable progress has been made in the epidemiology, molecular profiles, and risk factors for HCC. However, the 5-year survival rates of HCC patients in China are still less than 12% because many patients are not diagnosed until the cancer has developed to an advanced stage [3, 4]. In addition to the high molecular heterogeneity of HCC at three levels, namely interpatient, intertumoral, and intratumoral [5], it is imperative to explore the underlying molecular mechanism of HCC to identify novel effective prognostic factors that can be applied to diagnose HCC patients at an early stage and to reduce the mortality of HCC patients.
N6-Methyladenosine (m6A) regulates RNA metabolism, including splicing, export, degradation, and microRNA processing, through m6A methyltransferases (writer; the enzymes that deposit modification), demethylases (eraser; the enzymes that remove modification), and reader proteins (reader; the proteins that bind with the m6A sites of targeted RNA) [6–9]. The m6A modification is methylated at the N6 position on adenosine, which affects the localization, translocation efficiency, and variable splicing of mRNA [10]. It has been confirmed that m6A modification participates in regulating gene expression and plays important roles in many biological processes. Previous studies have reported the complex mechanisms of reversible m6A modification in human diseases [6, 11–13], even in cancer. Dynamic m6A levels have been demonstrated to participate in tumor development by modulating the expression of tumor-related genes in different types of tumor [7].
Recently, there is increasing evidence that dysregulation of m6A regulators may be involved in the progression of cancer, influencing the prognosis for patients with cancer. For instance, previous studies have shown that m6A methyltransferases, such as METTL14, are highly expressed and promote leukemogenesis in regulating targeted mRNAs through m6A modification in acute myeloid leukemia [14] as well as suppressing tumor progression by regulating the expression of related genes in a m6A modification manner [15]. In addition, YTH domain family member 2 (YTHDF2), a member of the YTHD family, suppresses the proliferation of HCC cells by destabilizing targeted mRNA [16]. These studies indicate that m6A serves important roles in the development of HCC. However, the association between m6A-related genes and HCC prognosis remains unclear.
In the present study, we obtained the RNA transcriptome and clinical data of HCC from The Cancer Genome Atlas (TCGA). We found three m6A regulators and 192 candidate m6A-related genes that were differentially expressed in different stages of HCC. We then analyzed the relationship between the expression of m6A candidate genes and progression in HCC patients. We finally selected 19 m6A-related genes to construct a risk model based on the risk score and explored the function of m6A-related genes in HCC development and prognosis. Our results provide new ideas for the integrated roles of m6A regulators and m6A-related genes in HCC.
Materials and methods
Data collection
RNA-seq and clinical data
We performed an analysis on TCGA level 3 RNA-seq transcriptome data. The FPKM data of HCC RNA-seq were downloaded from TCGA database along with the clinical data of the corresponding samples. In addition, we downloaded gene annotation files from Genecode. We selected samples that had both pathological characteristics (including stages I, II, III, and IV) and RNA-seq expression data. The epidemiological characteristics of HCC patients are presented in Table 1. We also downloaded the FPKM data for the HCC RNA-seq and the clinical data for the LIHC samples from the International Cancer Genome Consortium (ICGC) database.
Table 1 Clinicopathological Characteristics of LIHC Patients (n = 374) from TCGA Database
| Age | |
| ≤60 years | 260 (69.52) |
| >60 years | 113 (30.21) |
| Not reported | 1 (0.27) |
| Sex | |
| Female | 121 (32.35) |
| Male | 253 (67.65) |
| Pathological stage | |
| Not reported | 24 (6.42) |
| Stage I | 173 (46.26) |
| Stage II | 87 (23.26) |
| Stage III | 85 (22.73) |
| Stage IV | 5 (1.34) |
| Survival time | |
| ≤3 years | 105 (28.07) |
| >3 years | 25 (6.68) |
| Alive or not reported | 244 (65.24) |
| Survival status | |
| Alive | 243 (64.97) |
| Dead | 130 (34.76) |
| Not reported | 1 (0.27) |
Values are presented as n (%).
M6A regulators
We collected 21 m6A regulators from the literature [6, 17, 18], and these regulators are presented in Table S1. In addition, we downloaded LIHC m6A-related genes from the m6Avar website (http://m6avar.renlab.org/) [19], and 1258 m6A-related genes of HCC were obtained.
Integration analysis of differentially expressed m6A-related genes in HCC
After selecting liver cancer samples that had both pathological characteristics and RNA-seq data, we applied the gene annotation file to construct a profile of m6A-related genes in liver cancer (FPKM data). According to the stage standard for LIHC, these liver cancer samples were divided into 4 groups: stage I, stage II, stage III, and stage IV. We compared the expression values of the ICGC and TCGA data between different stages of HCC using one-way analysis of variance (p < 0.05). And then the selected differentially expressed genes were further used as the m6A candidate gene sets. Finally, a heatmap was constructed to visualize the differences.
Consensus clustering analysis
To elucidate the biological characteristics of m6A regulators in LIHC, we used the “ConsensusClusterPlus” package in R to perform an unsupervised cluster analysis on the samples [20]. All tumor samples corresponding to the candidate m6A-related genes were classified into two subgroups: KM1 and KM2.
Integration analysis of pathological characteristics and survival time of the KM1 and KM2 subgroups
We used a t-test to analyze the distribution of age between the KM1 and KM2 subgroups. In addition, we analyzed the difference in the World Health Organization (WHO) classification via the chi-square test and utilized the Cox regression model to explore the survival time difference between the KM1 and KM2 subgroups.
Interaction analysis among m6A candidate gene sets in HCC
The STRING database was used to explore the protein–protein interactions (PPIs) among m6A candidate genes. We used Spearman’s correlation analysis to explore the relationship among different m6A candidate genes.
Identification and enrichment analysis of differentially expressed genes between the KM1 and KM2 subgroups
After constructing the m6A candidate gene expression profiles of KM1 and KM2, we performed principal component analysis (PCA) to analyze the differentially expressed genes. We then used DAVID 6.759 to conduct functional annotation and enrichment analysis for the differentially expressed genes.
COX regression analysis and calculation
To determine the prognostic value of m6A candidate genes in HCC, a risk signature was developed for the dataset using univariate Cox regression analysis. Next, we filtered several important factor genes as prognostic genes of liver cancer. The risk score was calculated using the following equation:
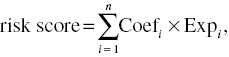
where Coefi is the coefficient and Expi is the expression of prognostic genes in each liver cancer sample. According to the median risk score, we divided the samples into low- and high-risk groups.
Risk scoring for prognosis and pathological characteristic prediction
We applied the LASSO regression algorithm (“glmnet” package in R) to TCGA liver cancer samples and obtained the risk score of 98 prognostic genes in each TCGA liver cancer sample. The 98 genes were then used to establish risk characteristics (LASSO automatically selected the best genes as characteristics) and the 1- to 5-year survival outcome of the HCC patients, and the grouping results for KM1 and KM2 were used as the predicted outcome. The predictive efficiency was then estimated by receiving operator characteristic (ROC) curves using the R software “survival” package.
Similarly, we applied the LASSO regression algorithm (“glmnet” package in R) to the ICGC liver cancer samples and obtained the risk score of 98 prognostic genes in each ICGC liver cancer sample. The 98 genes were then used to establish risk characteristics (LASSO automatically selected the best genes as characteristics), and the prognostic outcome was used as the predicted outcome. The predictive efficiency was then estimated by ROC curves with the “survival” package in R. The results indicated that the risk score provided predictors of prognostic outcome for cancer patients.
Analysis of differences in immune infiltration between low- and high-risk score groups
CIBERSORT provided a method to characterize cell composition based on gene expression profiles of complex tissues. The “LM22” white blood cell characteristic gene matrix composed of 547 genes was used to distinguish 22 immune cell types. In the present study, we used the results of the CIBERSORT evaluation published by Thorsson et al. [21]. We used the proportion of immune cells in samples from the low- and high-risk groups of HCC for statistical testing to analyze the difference in immune infiltration between the two subgroups.
Integration analysis of mutation differences and correlation analysis of risk score with matrix, immune score, and mRNAsi dryness index
We downloaded the liver cancer SNV data from TCGA and used the “maftools” package in R to display the distribution of mutations in the two subgroups. Based on the expression profile, we used the “estimate” package in R to evaluate the matrix score and immune score of the samples. According to the results of the tumor stemness index published by Malta et al. [22], the stemness index of liver cancer was extracted. The correlation of the immune score, stromal score, or mRNAsi with the two risk groups was then evaluated based on Pearson’s correlation coefficient analysis.
Results
Identification of m6A methylation candidate genes in HCC
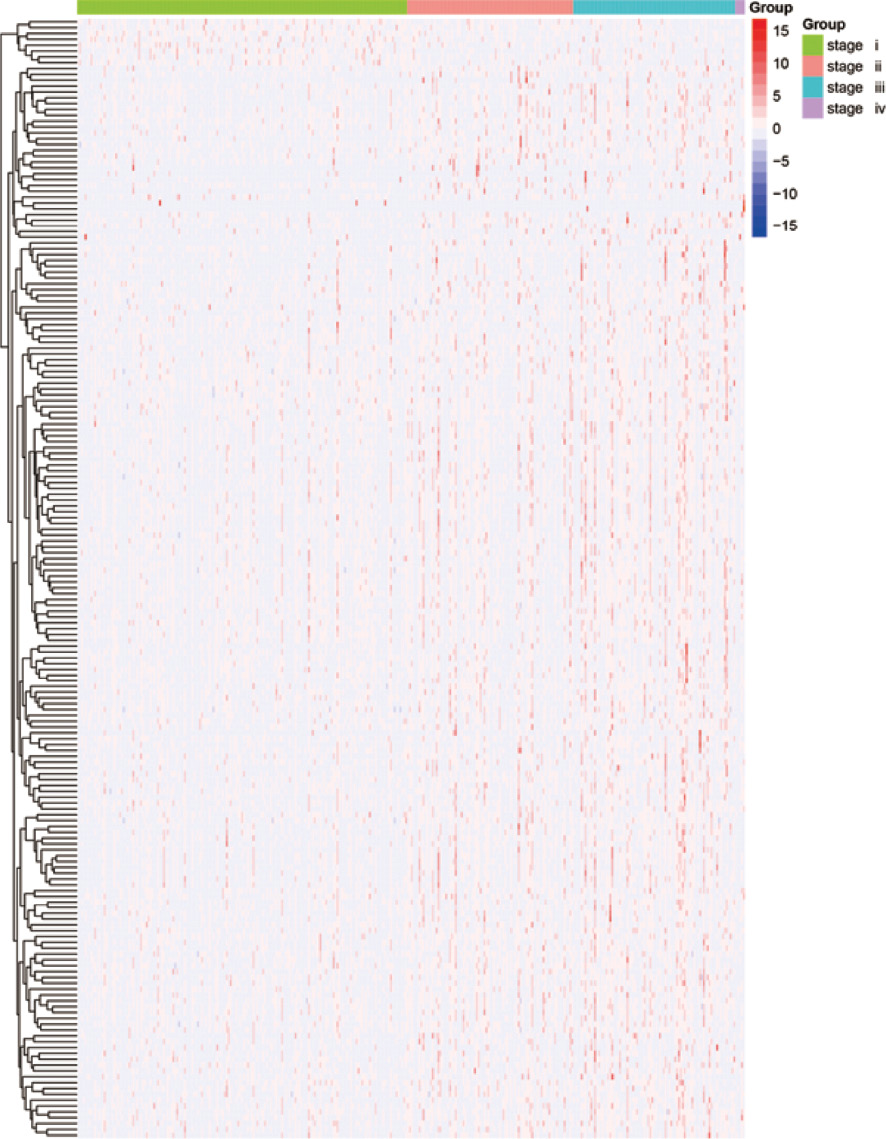
We obtained liver cancer samples that included both pathological characteristics and RNA-seq expression data. Based on the liver cancer stage, these samples were divided into 4 subgroups: stage I, stage II, stage III, and stage IV. After overlapping 1258 m6A-related genes of LIHC, we obtained differentially expressed genes (p < 0.05) among the four sample types by variance analysis. Finally, 192 differentially expressed genes were screened out as the m6A candidate gene set (Table S2). Notably, three of the m6A regulators, YTH domain-containing 1 (YTHDC1), YTH N6-methyladenosine RNA-binding protein 1 (YTHDF1), and YTH N6-methyladenosine RNA-binding protein 2 (YTHDF2), were expressed differently in the four subgroups. We then used the 192 m6A candidate genes to construct an expression heatmap. As shown in Figure 1, there was a significant difference among the expression levels of m6A candidate genes in different stages of HCC.
Figure 1 Expression of 192 m6A RNA methylation-related genes at different stages (I-IV) of hepatocellular carcinoma. The depth of red represents the level of high expression, and the depth of blue represents the level of low expression. m6A, N6-methyladenosine.
Consensus clustering analysis
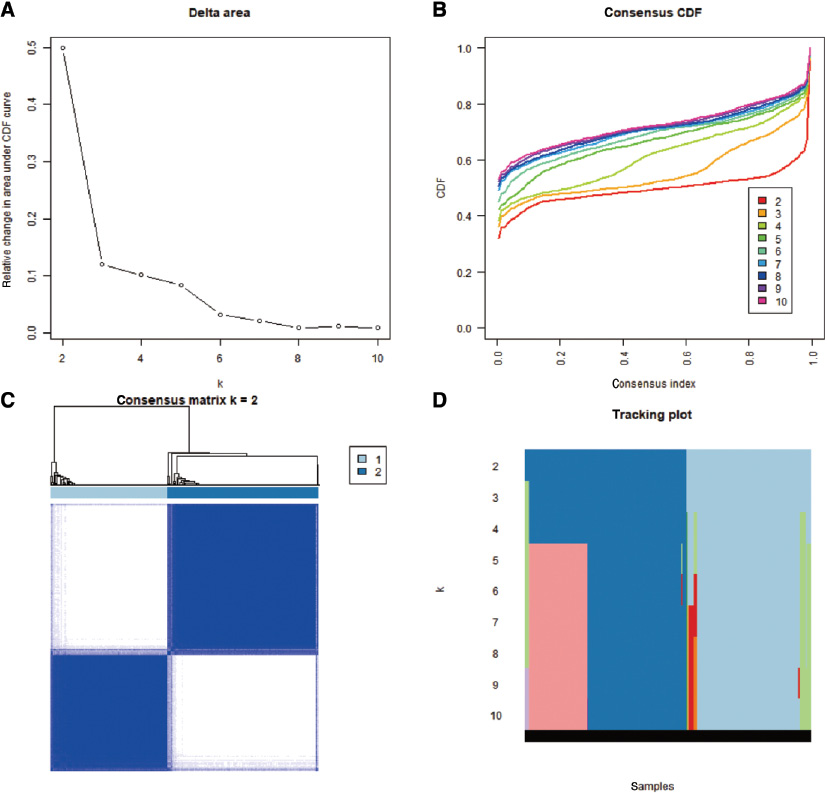
We performed consistent cluster analysis of HCC patients according to the expression of m6A regulators from TCGA. To explore the roles of m6A candidate genes in HCC, we used the expression levels of the 192 m6A candidate genes as feature vectors, and we then divided the HCC samples into different subgroups using the “Consensus Cluster Plus” package in R. In a clustering range from 2 to 10, we obtained a relatively clustering stable optimal k value (2) with TCGA data. The specific grouping conditions are presented in Table S3, and the conditions of sample-consistent clustering are shown in Figure 2. The consistent cluster cumulative distribution function (CDF) for k = 2–10 is presented in Figure 2A. The relative change in the area under the CDF curve is presented in Figure 2B; meanwhile, Figure 2C shows the visualization of the consistent clustering matrix between groups at k = 2. The distribution of the sample when k was between 2 and 10 is shown in Figure 2D. We named the subgroups KM1 and KM2.
Figure 2 Consistent cluster analysis of hepatocellular carcinoma. (A) The consistent cluster cumulative distribution function (CDF) when k is between 2 and 10. (B) The relative change in the area under the CDF curve when k ranges from 2 to 10. (C) Visualization of the consistent clustering matrix between groups at k = 2. (D) The distribution of the sample when k is between 2 and 10.
Integration analysis of pathological characteristics and survival time of the KM1 and KM2 subgroups
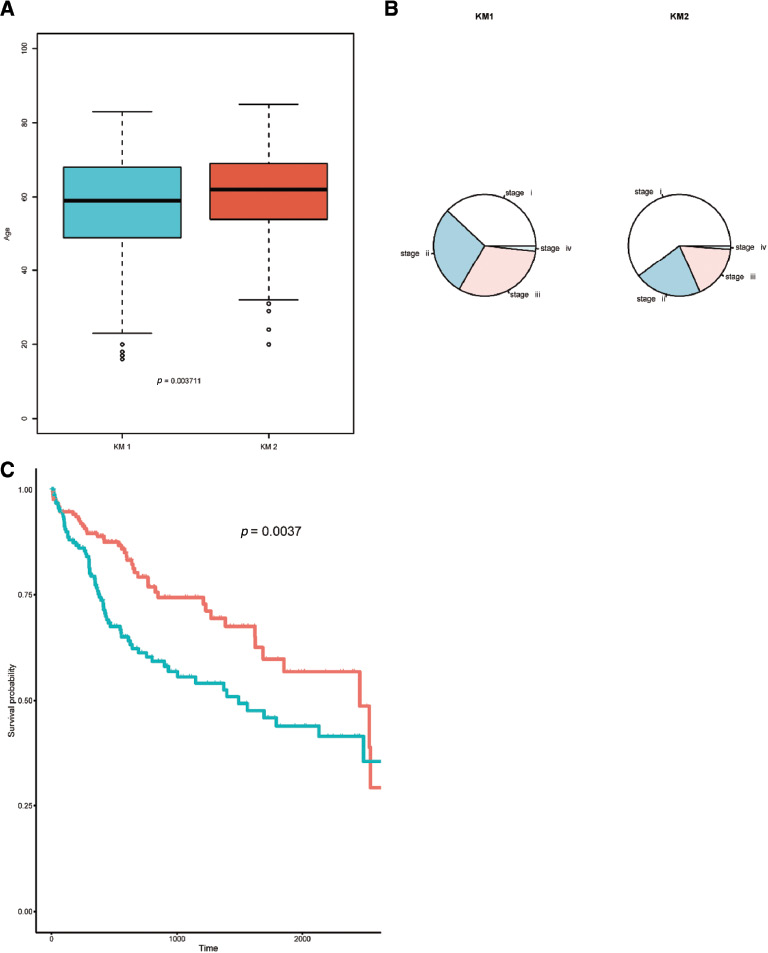
We extracted the clinical information from the two subgroups (Table S4) and performed a t-test to analyze the age distribution of the KM1 and KM2 subgroups. There was a significant difference in age between the two subgroups (p = 0.004) (Figure 3A). We extracted the samples containing stage data and analyzed the differences in the WHO staging of the two subgroups via the chi-square test. The WHO classifications were significantly different between the two subgroups (p < 0.001). The WHO classification pie chart of the two subgroups is shown in Figure 3B. We also extracted samples containing survival time from the KM1 and KM2 subgroups, and Cox regression was then used to explore the survival time between the two subgroups. Figure 3C shows the KM survival curve, and the results indicated a significant difference (p = 0.004) in survival time between the two subgroups.
Figure 3 Comparative analysis of pathological characteristics and survival time between the two subgroups. (A) Box plot of age between the two subgroups of liver cancer. There was a significant difference in age (p < 0.05). (B) WHO classification pie chart between two subgroups of liver cancer. There was a significant difference in WHO classification (p < 0.001). (C) Survival curves of the two subgroups in liver cancer. There was a significant difference in survival time (p < 0.01).
Interaction analysis among m6A candidate gene sets in HCC
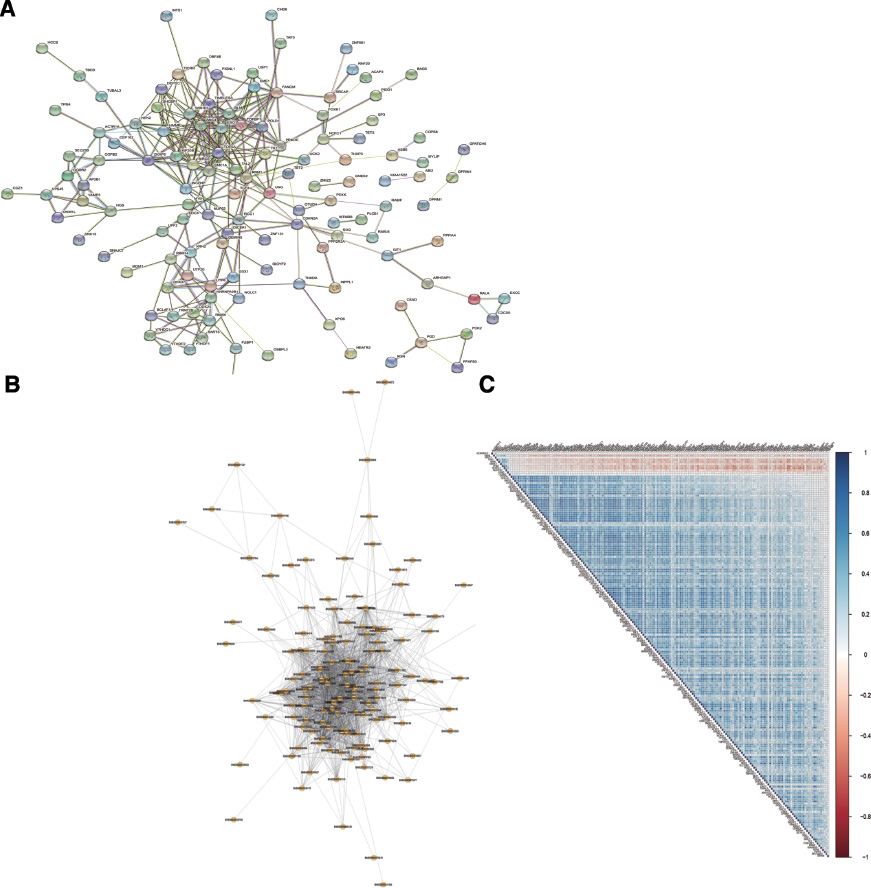
To explore the association network of functional proteins among these 192 m6A candidate genes, we generated a PPI network of the candidate gene set using STRING (Figure 4A). We then calculated the Spearman’s correlation between every two m6A candidate genes based on the m6A candidate gene expression profile. In this study, we selected genes with cor > 0.6 and p < 0.05 (Table S5) to construct a correlation network (Figure 4B). Spearman’s analysis was used to analyze the correlation of the expression levels among the factors. The correlation coefficient threshold of the expression level was |r| ≥ 0.3, and the p value of the rank sum test was p < 0.01. As shown in Figure 4C, the correlation of all 192 genes was analyzed. In Figure 4C, blue represents a positive correlation, whereas red represents a negative correlation.
Figure 4 Interaction analysis among m6A candidate gene sets in hepatocellular carcinoma. (A) Protein-protein interaction (PPI) network of m6A candidate genes. (B) The coexpression network of m6A candidate genes. (C) Correlation of expression from 192 liver cancer-related genes. Blue represents a positive correlation, and red represents a negative correlation.
Identification and enrichment analysis of differentially expressed genes between KM1 and KM2
We explored the expression differences of samples between the two subgroups via PCA. The results (Figure 5A and B) suggested that the RNA expression of the two subgroups was specific. However, there were still some overlapping areas in the two subgroups, suggesting that both subgroups may share some commonality.
Figure 5 Identification and functional enrichment analysis of differentially expressed genes between KM1 and KM2. (A) Principal component change range. (B) Principal component analysis of the KM1 and KM2 subgroups. (C) GO functional enrichment of differentially expressed genes between the KM1 and KM2 subgroups. (D) KEGG functional enrichment of differentially expressed genes between the KM1 and KM2 subgroups.
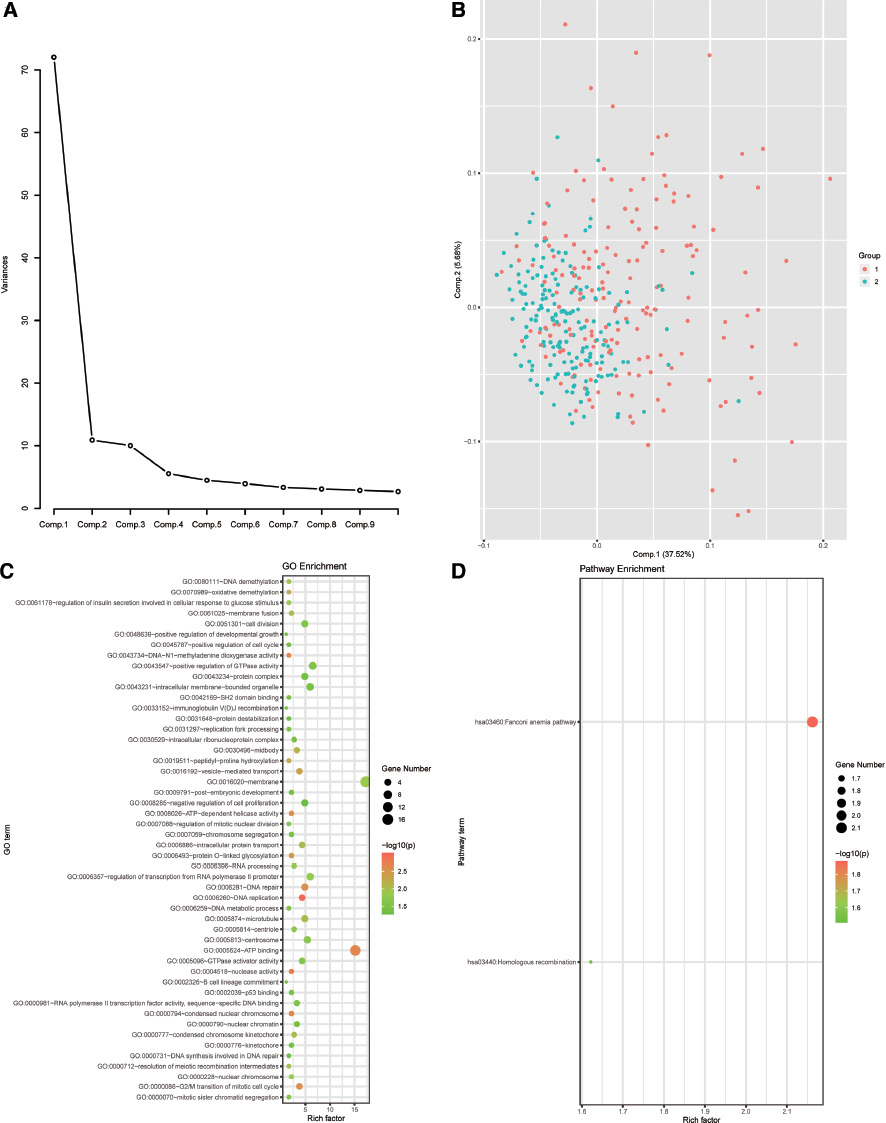
We then used a t-test to explore the differentially expressed genes between the two subgroups (p < 0.05) (Table S6). GO function and KEGG pathway enrichment analyses of the differentially expressed genes were performed with DAVID (Table S7 and Table S8). The results indicated that the differences between the two subgroups were mainly enriched in several GO functions, such as cell membrane-related lipids and proteins as well as intracellular energy protein binding. These genes were also enriched in pathways associated with Fanconi anemia and homologous recombination. These pathways were closely related to DNA repair, cross-linking, and precise repair of DNA double-strand breaks (Figure 5C and D).
Analysis of univariate Cox regression and prognostic value of m6A candidate genes
To explore the prognostic value of m6A candidate genes in HCC, we used the “survival” package in R to perform univariate Cox regression analysis. A total of 343 samples remained after removing those samples without survival-related information. The results indicated that among 192 candidate genes, the expression of 98 candidate genes correlated with the survival of HCC (p < 0.05).
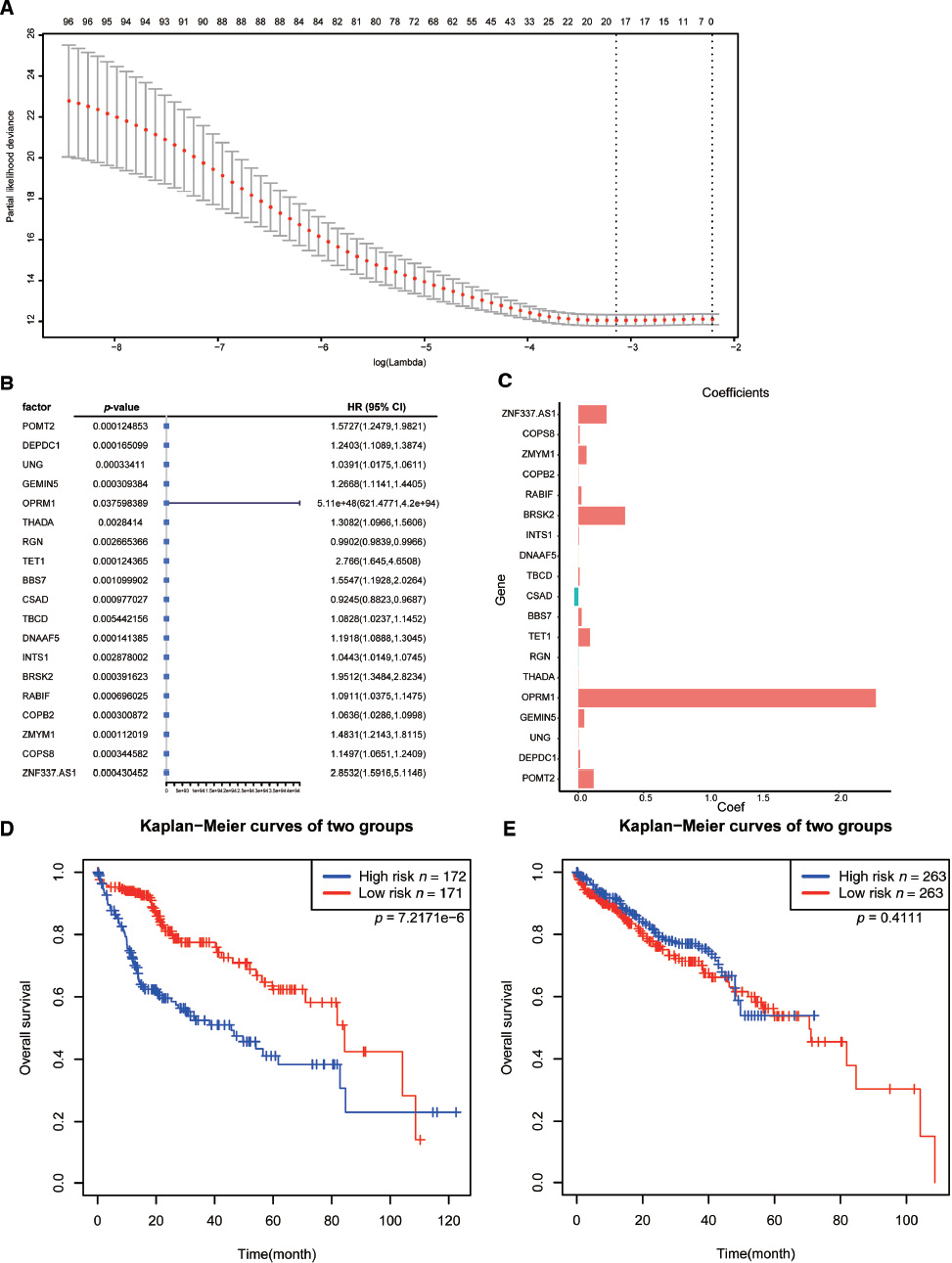
To further predict the prognosis of m6A regulatory genes in liver cancer, we analyzed 98 gene sets with the LASSO regression analysis algorithm. As a result, 19 genes were obtained to construct risk characteristics. Of these 19 genes for the prognostic model, POMT2, DEPDC1, UNG, GEMIN5, OPRM1, THADA, TET1, BBS7, TBCD, DNAAF5, INTS1, BRSK2, RABIF, COPB2, ZMYM1, COPS8, and ZNF337.AS1, were related to poor overall survival, whereas regucalcin (RGN) and cysteine sulfinic acid decarboxylase (CSAD) were related to good overall survival in LIHC (Figure 6A–C). We then used the regression coefficient to calculate risk scores. To clarify the prognostic efficacy of these characteristics, we divided the samples into two subgroups according to the median value of risk scores. As shown in Figure 6D, the high- and low-risk groups had significant differences in survival (p < 0.001). To validate the stability of the risk scores, we utilized the ICGC data to evaluate the efficacy of these features. The results suggested that 17 of the 19 candidate genes with significant survival existed in the ICGC data, and the remaining two genes had no expression (as indicated by “NA”). We used the expression of the 17 genes for survival analysis, which indicated that there was no significant difference in the survival results (p = 0.411) (Figure 6E).
Figure 6 Nineteen survival-related m6A RNA methylation-related genes were screened. (A) Least absolute shrinkage and selection operator (LASSO) regression analysis was used to screen 98 m6A RNA methylation-related genes. (B, C) The 95% confidence intervals (CIs) obtained by univariate Cox regression and the coefficients calculated with multivariate Cox regression using funnel plots. (D, E) Kaplan-Meier curves of the two groups from TCGA and ICGC.
Prognostic value of m6A candidate genes with different clinicopathological features and clinical outcomes for HCC
We analyzed the differences in age, sex and stage of the patients in the two subgroups. The results showed that sex (p = 0.008) and stage (p < 0.001) in the high-risk group were significantly different from those in the low-risk group, whereas there was no obvious difference in age between the two groups (p = 0.178). We also explored the correlation between risk scores and different clinical characteristics. The results indicated significant correlations between risk score and sex (p = 0.013) and stage (p < 0.001). However, the risk scores were not significantly correlated to age (p = 0.102). The ability of the risk score to be used for predicting the 1- and 5-year survival rate of HCC patients is presented in Figure 7A–E. The ROC curve indicated that the risk score could be used to predict the survival of HCC patients at 1 to 3 years [areas under the curve (AUC) >0.7]. As shown in Figure 7F and G, we generated a nomogram via multivariate Cox regression analysis to predict the probability of 1-, 2-, 3-, 4-, and 5-year OS from data containing significant factors, such as age, stage, and risk score. The total points of the samples under certain conditions in the nomogram allowed an understanding of the survival situation and prediction of the 1- to 5-year survival rate of the samples. The accuracy of the prediction model was evaluated with the consistency index (C-index). The C-index was 0.718, which suggested that the accuracy was good.
Figure 7 Analysis of prognostic and clinical characteristics. (A) Risk score predicting the 1-year survival rate of liver cancer patients. (B) Risk score predicting the 2-year survival rate of liver cancer patients. (C) Risk score predicting the 3-year survival rate of liver cancer patients. (D) Risk score predicting the 4-year survival rate of liver cancer patients. (E) Risk score predicting the 5-year survival rate of liver cancer patients. (F) Nomogram composed of age, stage and risk score for the prediction of 1-year, 2-year, 3-year, 4-year and 5-year survival probability. (G) Calibration curve for the evaluation of the nomogram in predicting 1-year, 2-year, 3-year, 4-year and 5-year survival probability. (H) Relationship between risk score and survival time. In the survival time plot, a blue circle indicates that the sample status is alive, and the red circle indicates that the sample status is dead. (I) Heatmap presenting the expression levels of 19 m6A RNA methylation-related genes in the low- and high-risk groups. The distribution of clinicopathological features was analyzed between the low- and high-risk groups.
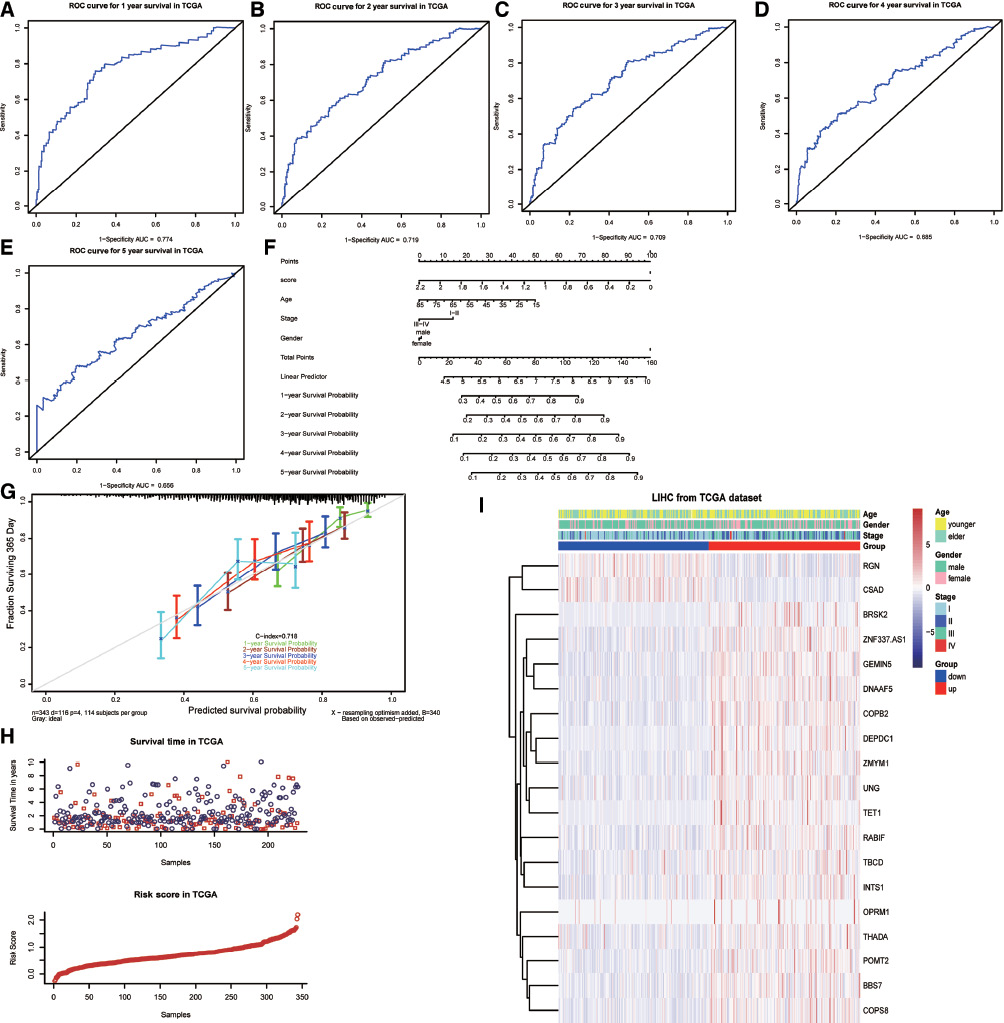
To explore whether these risk characteristics could function as novel prognostic risk factors, we studied the relationship among the risk score, clinicopathological characteristics (including age, stage and sex) of TCGA data samples, and survival. The results showed that only the risk score was significantly related to survival (p < 0.001) (Table S9 and Figure 7H). The genetic status and clinical features with significant survival in the two subgroups are shown in the heatmap (Figure 7I).
Analysis of differences in immune infiltration of the two subgroups
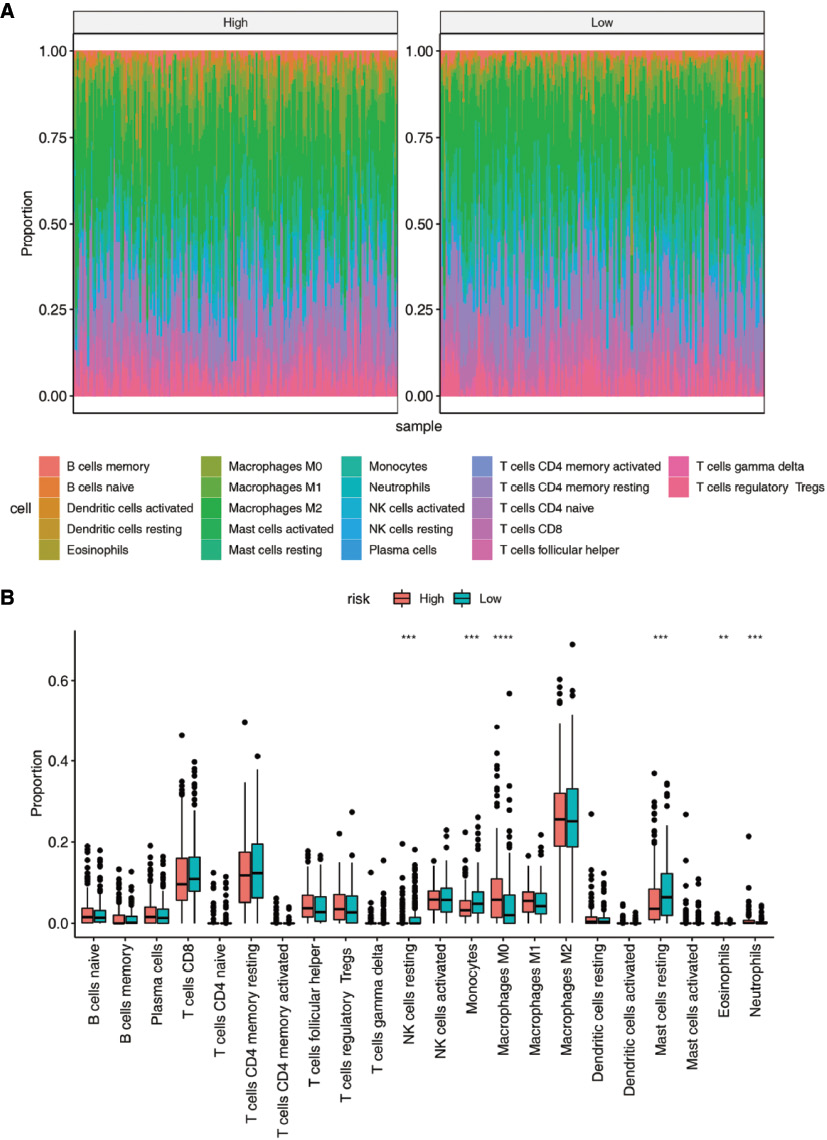
Growing evidence has demonstrated that the tumor microenvironment greatly influences the development of various tumors, such as HCC [23]. To explore the underlying mechanism of these m6A regulators in HCC, we further evaluated the abundance of different immune cells from high- and low-risk groups via the “CIBERSORT” package in R. The integrative distributions of the fractions among 22 immune cell types from the two groups are shown in Figure 8A. In the low-risk group, the immune infiltration of monocytes, NK cells, and mast cells was higher than that in the high-risk group, whereas there were more M0 macrophages in the high-risk group than in the low-risk group (Figure 8B).
Figure 8 Analysis of differences in immune infiltration between high- and low-risk score groups in HCC. (A) Ratio distribution of immune cells. (B) Differences in immune infiltration between the high- and low-risk groups in HCC.
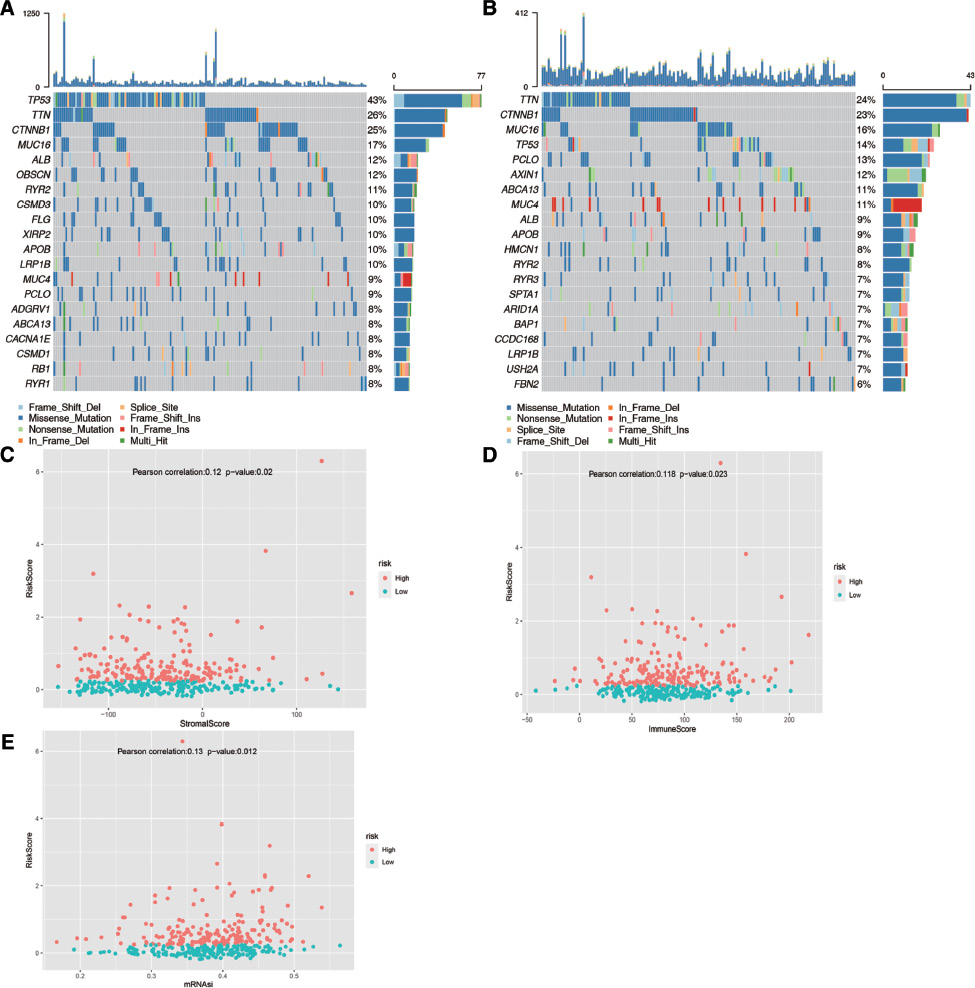
Analysis of mutation differences between the two subgroups and correlation analysis of risk score with matrix, immune score, and mRNAsi dryness index
In addition, we performed mutation analysis. The landscape of mutation information of each gene in each sample from different groups (high- and low-groups) is exhibited in Figure 9A and B. The various colors in the waterfall plot correspond to different mutation types, and the pictures present the top 20 mutated genes in HCC with ranked percentages in the two subgroups. We found that the gene with the highest mutation frequency was TP53 (43%) in the high-risk group and TTN (24%) in the low-risk group. Moreover, we analyzed the relationship among the risk score and the stromal score, immune score and mRNAsi score via Pearson’s correlation analysis. The results revealed that there were significant differences between the risk score and the stromal score, immune score, and mRNAsi score (all p < 0.05) (Figure 9C–E).
Figure 9 Landscape of mutation profiles in HCC samples. (A) High and (B) low mutation information of each gene in each sample is shown in the waterfall plot, in which various colors with annotations at the bottom represent the different mutation types. The bar plot above the legend shows the mutation burden. Significant differences among the stromal score (C), immune score (D), mRNAsi score (E) and risk score (all p < 0.05).
Discussion
HCC is the fourth leading cause of cancer-related deaths and the sixth most common cancer worldwide. Although there has been a large advancement in the therapeutic management of HCC, patients with HCC still have poor prognosis [24]. Accumulating evidence has shown that m6A modification is linked to tumor proliferation [25], tumorigenesis, differentiation, invasion [26], and metastasis [27], and m6A modification acts both as an oncogene and an anti-oncogene in tumors. Thus, identifying m6A regulators and m6A-related genes in HCC may offer novel insight into valuable therapeutic targets.
The influence of METTL3 and METTL14 on HCC development and progression has been widely reported in previous studies [6], whereas the function of m6A-related genes is not fully elucidated. Therefore, we explored the key genes from the 1258 candidate m6A-related genes and then analyzed their correlation with the clinicopathological characteristics of HCC patients. Our study obtained 192 m6A-related candidate genes that were differentially expressed in different pathological stages of HCC patients. In addition, we found that the expression of three m6A regulators, YTHDF1, YTHDF2, and YTHDC1, was linked to the prognosis of HCC.
As RNA-binding proteins of the YTH domain family, YTHDF1 and YTHDF2 were significantly upregulated in various cancers, including HCC, which affected the cell cycle and metabolism of tumor cells. It has been reported that high YTHDF1 expression implies poor prognosis of HCC patients [28]. In addition, a previous study has found that YTHDF2 may act as a tumor suppressor to inhibit cell proliferation in HCC [16]. Recent evidence has shown that YTHDF1 may act as a prognostic factor and has an opportunity to be involved in the treatment of colon cancer patients [29]. Silencing of YTHDF2 enhances the inflammation, metastasis and angiogenesis of HCC [30]. Study found that the expression of YTHDF2 was related to infiltration of immune cells and their marker genes. In addition,YTHDF2 may have the potential effect in regulating polarization of tumor-associated macrophages, such as T-cell exhaustion, and activating T-regulatory cells [31]. YTHDC1 regulates the splicing of exons by binding to splicing factors [32]. Our study demonstrated that the expression of YTHDF1, YTHDF2, and YTHDC1 was significantly different at the four pathological stages of HCC. Therefore, the expression of m6A regulators may be strongly related to the clinicopathological features of HCC patients.
In addition, we obtained two HCC subtypes (KM1 and KM2) via consensus clustering analysis. The age, WHO classification, and survival time differed between the KM1 and KM2 subtypes (all p < 0.05) of HCC patients. In addition, we explored the differentially expressed genes between the two subgroups followed by GO function and KEGG pathway enrichment analysis. The results indicated that these genes were enriched in pathways associated with the malignant progression of HCC. However, the specific molecular mechanisms still require further study.
To explore the prognostic roles of m6A candidate genes in HCC, we used univariate analysis and LASSO regression to construct a 19-gene risk model. Among these 19 prognosis-related genes, the eight top genes were ZMYM1, BRSK2, POMT2, BBS7, TET1, THADA, GEMIN5, and DEPDC1. Previous studies have revealed that DEP domain-containing 1 (DEPDC1) is an oncogene that is upregulated and drives cell proliferation, invasion, and angiogenesis [33] but suppresses sensitivity to chemotherapy in HCC [34]. The gem nuclear organelle-associated protein 5 (GEMIN5) RNA-binding protein binds to the ribosome and is involved in global translation [35], and this alteration contributes to alternative splicing and tumor cell motility [36]. Ten–eleven translocation 1 (TET1), a widely reported DNA demethylation protein, has been reported to be expressed at low levels in HCC tissues, which facilitates HCC cell invasion by reducing the inhibitory effect of miR-494-induced HCC cell invasiveness [37]. This is consistent with the results in our study. Thus, our study provides a reference for these genes for further research.
We then divided the samples from TCGC database into high- and low-risk groups based on the median risk scores. Kaplan–Meier analysis indicated that the survival of the two subgroups was obviously distinguished. In addition, the AUCs of 1 year to 3 years among HCC patients were greater than 0.7, indicating that our risk model has the potential to predict the survival of HCC patients. We also explored the correlation among the risk score, clinicopathological characteristics (including age, stage, and sex) and survival for the samples from TCGA data. The results revealed that only the risk score was significantly related to survival, indicating the prognostic value of the risk model for HCC.
By analyzing mutation differences between high- and low-risk score groups, we found that TP53 and TTN had the highest mutation frequency in the high-risk and low-risk HCC patient groups, respectively. TP53, which has been named “the guardian of the genome,” is an important tumor-suppressor gene. The mutation in TP53 and the resultant inactivation of p53 cause the evasion of rapid tumor progression and tumor cell death [38]. In breast cancer, the mutation of TP53 was the most prevalent in patients with the basal/triple negative subtype. Jessica et al. reported that TP53 is the next most prevalent mutation, affecting 25% to 30% of HCC patients [39]. Another study has reported that TP53 pathways and regulatory miRs may be used as therapeutic targets in HCC [40]. In our study, we found that TP53 had the highest mutation frequency in the high-risk HCC patient group, which agreed with existing research and suggested the use of TP53 to assess the risk of liver cancer.
TTN, the longest-known coding gene, has been reported to be frequently mutated in some tumors, including lung adenocarcinoma and colon adenocarcinoma. It has been reported that patients with lung squamous cell carcinoma have favorable overall survival benefits from the TTN mutant type [41]. Jia et al. [42] reported that the mutation rate of TTN is 29.68% in solid tumors, and they divided tumor mutation burden into high and low groups, representing different clinical responses to immune checkpoint blockade monotherapy. However, another study has found that spontaneous mutation in the TTN gene represents a high tumor mutation burden [43]. In our study, we found that TTN had the highest mutation frequency in the low-risk HCC patient groups. Thus, we hypothesize that m6A RNA methylation regulated by TP53 and TTN may play an important role in regulating tumor proliferation and metastasis, which further influences the prognosis of HCC patients.
Conclusion
We analyzed the expression, PPI, and prognostic value of m6A-related genes in HCC. Our results indicated an close relationship between the expression of m6A-related genes and clinicopathological characteristics in HCC. We identified several m6A-related genes that may serve as novel prognostic markers in HCC patients. Our study provides integrated evidence for exploring the function of m6A in HCC and further development of therapeutics.
Data availability
All datasets used in our study are publicly available. The data can be downloaded from The Cancer Genome Atlas (TCGA), Gencode, International Cancer Genome Consortium Databases (ICGC), m6Avar database, STRING database, and CIBERSORT (http://cancergenome.nih.gov/, https://www.gencodegenes.org/, https://icgc.org/, http://m6avar.renlab.org/, http://string-db.org/, https://cibersort.stanford.edu/).
Conflicts of Interest
The authors declare that they have no competing interests.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (grant numbers 81870878, 81971057, and 81902490), Guangdong Natural Science Foundation for Distinguished Young Scholars (grant number 2019B151502010), and Sun Yat-sen University Training Project Foundation for Young Teachers (19ykpy183).
Supplementary Tables
Click here to download Supplementary Tables.
References
- Yang JD, Hainaut P, Gores GJ, Amadou A, Plymoth A, et al. A global view of hepatocellular carcinoma: trends, risk, prevention and management. Nat Rev Gastroenterol Hepatol 2019;16:589-604. [PMID: 31439937 DOI: 10.1038/s41575-019-0186-y]
- Wong C-M, Tsang FH-C, Ng IO-L. Non-coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol 2018;15:137-51. [PMID: 29317776 DOI: 10.1038/nrgastro.2017.169]
- Zheng R, Qu C, Zhang S, Zeng H, Sun K, et al. Liver cancer incidence and mortality in China: temporal trends and projections to 2030. Chin J Cancer Res 2018;30:571-9. [PMID: 30700925 DOI: 10.21147/j.issn.1000-9604.2018.06.01]
- Roxburgh P, Evans TR. Systemic therapy of hepatocellular carcinoma: are we making progress? Adv Ther 2008;25:1089-104. [PMID: 18972075 DOI: 10.1007/s12325-008-0113-z]
- Craig AJ, von Felden J, Garcia-Lezana T, Sarcognato S, Villanueva A. Tumour evolution in hepatocellular carcinoma. Nat Rev Gastroenterol Hepatol 2020;17:139-52. [PMID: 31792430 DOI: 10.1038/s41575-019-0229-4]
- Batista PJ. The RNA modification N-methyladenosine and its implications in human disease. Genom Proteom Bioinform 2017;15:154-63. [PMID: 28533023 DOI: 10.1016/j.gpb.2017.03.002]
- He L, Li H, Wu A, Peng Y, Shu G, et al. Functions of N6-methyladenosine and its role in cancer. Mol Cancer 2019;18:176. [PMID: 31801551 DOI: 10.1186/s12943-019-1109-9]
- Liu N, Zhou KI, Parisien M, Dai Q, Diatchenko L, et al. N6-methyladenosine alters RNA structure to regulate binding of a low-complexity protein. Nucl Acids Res 2017;45:6051-63. [PMID: 28334903 DOI: 10.1093/nar/gkx141]
- Liu Q, Gregory RI. RNAmod: an integrated system for the annotation of mRNA modifications. Nucl Acids Res 2019;47:W548-55. [PMID: 31147718 DOI: 10.1093/nar/gkz479]
- Fu Y, Dominissini D, Rechavi G, He C. Gene expression regulation mediated through reversible m6A RNA methylation. Nat Rev Genet 2014;15:293-306. [PMID: 24662220 DOI: 10.1038/nrg3724]
- Nilsen TW. Molecular biology. Internal mRNA methylation finally finds functions. Science 2014;343:1207-8. [PMID: 24626918 DOI: 10.1126/science.1249340]
- Roundtree IA, Evans ME, Pan T, He C. Dynamic RNA modifications in gene expression regulation. Cell 2017;169:1187-200. [PMID: 28622506 DOI: 10.1016/j.cell.2017.05.045]
- Zhao BS, Roundtree IA, He C. Post-transcriptional gene regulation by mRNA modifications. Nat Rev Mol Cell Biol 2017;18:31-42. [PMID: 27808276 DOI: 10.1038/nrm.2016.132j]
- Weng H, Huang H, Wu H, Qin X, Zhao BS, et al. METTL14 inhibits hematopoietic stem/progenitor differentiation and promotes leukemogenesis via mRNA m(6)A modification. Cell Stem Cell 2018;22:191-205.e9. [PMID: 29290617 DOI: 10.1016/j.stem.2017.11.016]
- Zang YX, Li S, Ruan F, Liu ZC, Jie Y. Tacrolimus dye drop treatment for the management of early post-operative intraocular inflammation after therapeutic keratoplasty for severe infectious keratitis. Exp Ther Med 2020;20:3260-8. [PMID: 32855696 DOI: 10.3892/etm.2020.9057]
- Zhong L, Liao D, Zhang M, Zeng C, Li X, et al. YTHDF2 suppresses cell proliferation and growth via destabilizing the EGFR mRNA in hepatocellular carcinoma. Cancer Lett 2019;442:252-61. [PMID: 30423408 DOI: 10.1016/j.canlet.2018.11.006]
- Sun T, Wu R, Ming L. The role of m6A RNA methylation in cancer. Biomed Pharmacother 2019;112:108613. [PMID: 30784918 DOI: 10.1016/j.biopha.2019.108613]
- Yang Y, Hsu PJ, Chen Y-S, Yang Y-G. Dynamic transcriptomic mA decoration: writers, erasers, readers and functions in RNA metabolism. Cell Res 2018;28:616-24. [PMID: 29789545 DOI: 10.1038/s41422-018-0040-8]
- Zheng Y, Nie P, Peng D, He Z, Liu M, et al. m6AVar: a database of functional variants involved in m6A modification. Nucl Acids Res 2018;46(D1):D139-45. [PMID: 29036329 DOI: 10.1093/nar/gkx895]
- Wilkerson MD, Hayes DN. ConsensusClusterPlus: a class discovery tool with confidence assessments and item tracking. Bioinformatics 2010;26:1572-3. [PMID: 20427518 DOI: 10.1093/bioinformatics/btq170]
- Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, et al. The immune landscape of cancer. Immunity 2018;48:812-30.e14. [PMID: 31771653 DOI: 10.1186/s40880-019-0427-z]
- Malta TM, Sokolov A, Gentles AJ, Burzykowski T, Poisson L, et al. Machine learning identifies stemness features associated with oncogenic dedifferentiation. Cell 2018;173:338-54.e15. [PMID: 29625051 DOI: 10.1016/j.cell.2018.03.034]
- Lu C, Rong D, Zhang B, Zheng W, Wang X, et al. Current perspectives on the immunosuppressive tumor microenvironment in hepatocellular carcinoma: challenges and opportunities. Mol Cancer 2019;18:130. [PMID: 31464625 DOI: 10.1186/s12943-019-1047-6]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394-424. [PMID: 30207593 DOI: 10.3322/caac.21492]
- Liu J, Eckert MA, Harada BT, Liu S-M, Lu Z, et al. mA mRNA methylation regulates AKT activity to promote the proliferation and tumorigenicity of endometrial cancer. Nat Cell Biol 2018;20:1074-83. [PMID: 30154548 DOI: 10.1038/s41556-018-0174-4]
- Lin S, Choe J, Du P, Triboulet R, Gregory RI. The m(6)A methyltransferase METTL3 promotes translation in human cancer cells. Mol Cell 2016;62:335-45. [PMID: 27117702 DOI: 10.1016/j.molcel.2016.03.021]
- Ma J-Z, Yang F, Zhou C-C, Liu F, Yuan J-H, et al. METTL14 suppresses the metastatic potential of hepatocellular carcinoma by modulating N-methyladenosine-dependent primary microRNA processing. Hepatology 2017;65:529-43. [PMID: 27774652 DOI: 10.1002/hep.28885]
- Zhao X, Chen Y, Mao Q, Jiang X, Jiang W, et al. Overexpression of YTHDF1 is associated with poor prognosis in patients with hepatocellular carcinoma. Cancer Biomark A 2018;21:859-68. [PMID: 29439311 DOI: 10.3233/CBM-170791]
- Xu D, Shao J, Song H, Wang J. The YTH Domain family of N6-methyladenosine “readers” in the diagnosis and prognosis of colonic adenocarcinoma. BioMed Res Int 2020;2020:9502560. [PMID: 32596399 DOI: 10.1155/2020/9502560]
- Hou J, Zhang H, Liu J, Zhao Z, Wang J, et al. YTHDF2 reduction fuels inflammation and vascular abnormalization in hepatocellular carcinoma. Mol Cancer 2019;18:163. [PMID: 31735169 DOI: 10.1186/s12943-019-1082-3]
- Shao X-Y, Dong J, Zhang H, Wu Y-S, Zheng L. Systematic analyses of the role of the reader protein of -methyladenosine RNA methylation, YTH domain family 2, in liver hepatocellular carcinoma. Front Mol Biosci 2020;7:577460. [PMID: 33344502 DOI: 10.3389/fmolb.2020.577460]
- Kasowitz SD, Ma J, Anderson SJ, Leu NA, Xu Y, et al. Nuclear m6A reader YTHDC1 regulates alternative polyadenylation and splicing during mouse oocyte development. PLoS Genet 2018;14:e1007412. [PMID: 29799838 DOI: 10.1371/journal.pgen.1007412]
- Guo W, Li H, Liu H, Ma X, Yang S, et al. DEPDC1 drives hepatocellular carcinoma cell proliferation, invasion and angiogenesis by regulating the CCL20/CCR6 signaling pathway. Oncol Rep 2019;42:1075-89. [PMID: 31322256 DOI: 10.3892/or.2019.7221]
- Zhou C, Wang P, Tu M, Huang Y, Xiong F, et al. DEPDC1 promotes cell proliferation and suppresses sensitivity to chemotherapy in human hepatocellular carcinoma. Biosci Rep 2019;39:BSR20190946. [PMID: 31189746 DOI: 10.1042/BSR20190946]
- Francisco-Velilla R, Fernandez-Chamorro J, Ramajo J, Martinez-Salas E. The RNA-binding protein Gemin5 binds directly to the ribosome and regulates global translation. Nucl Acids Res 2016;44:8335-51. [PMID: 27507887 DOI: 10.1093/nar/gkw702]
- Lee JH, Horak CE, Khanna C, Meng Z, Yu LR, et al. Alterations in Gemin5 expression contribute to alternative mRNA splicing patterns and tumor cell motility. Cancer Res 2008;68:639-44. [PMID: 18245461 DOI: 10.1158/0008-5472.CAN-07-2632]
- Chuang KH, Whitney-Miller CL, Chu CY, Zhou Z, Dokus MK, et al. MicroRNA-494 is a master epigenetic regulator of multiple invasion-suppressor microRNAs by targeting ten eleven translocation 1 in invasive human hepatocellular carcinoma tumors. Hepatology (Baltimore, Md) 2015;62:466-80. [PMID: 25820676 DOI: 10.1002/hep.27816]
- Bykov VJN, Eriksson SE, Bianchi J, Wiman KG. Targeting mutant p53 for efficient cancer therapy. Nat Rev Cancer 2018;18:89-102. [PMID: 29242642 DOI: 10.1038/nrc.2017.109]
- Zucman-Rossi J, Villanueva A, Nault J-C, Llovet JM. Genetic landscape and biomarkers of hepatocellular carcinoma. Gastroenterology 2015;149:1226-39.e4. [PMID: 26099527 DOI: 10.1053/j.gastro.2015.05.061]
- Akula SM, Abrams SL, Steelman LS, Emma MR, Augello G, et al. RAS/RAF/MEK/ERK, PI3K/PTEN/AKT/mTORC1 and TP53 pathways and regulatory miRs as therapeutic targets in hepatocellular carcinoma. Exp Opin Ther Targets 2019;23:915-29. [PMID: 31657972 DOI: 10.1080/14728222.2019.1685501]
- Cheng X, Yin H, Fu J, Chen C, An J, et al. Aggregate analysis based on TCGA:TTN missense mutation correlates with favorable prognosis in lung squamous cell carcinoma. J Cancer Res Clin Oncol 2019;145:1027-35. [PMID: 30810839 DOI: 10.1007/s00432-019-02861-y]
- Jia Q, Wang J, He N, He J, Zhu B. Titin mutation associated with responsiveness to checkpoint blockades in solid tumors. JCI Insight 2019;4:e127901. [PMID: 31092729 DOI: 10.1172/jci.insight.127901]
- Oh J-H, Jang SJ, Kim J, Sohn I, Lee J-Y, et al. Spontaneous mutations in the single gene represent high tumor mutation burden. NPJ Genom Med 2020;5:33. [PMID: 33436630 DOI: 10.1038/s41525-019-0107-6]